
Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
Alkaptonurie ist eine angeborene Enzymabnormalität
Facharzt des Artikels

Zuletzt überprüft: 04.07.2025
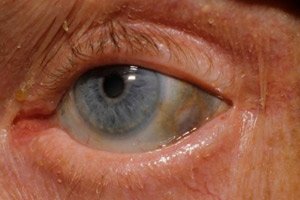
Bei einer der sehr seltenen Stoffwechselerkrankungen, der Alkaptonurie, handelt es sich um angeborene Anomalien im Stoffwechsel der Aminosäure Tyrosin.
Dieses Syndrom kann auch als Homogentisatoxidasemangel, Homogentisinurie, hereditäre Ochronose oder Schwarzurinkrankheit bezeichnet werden.[ 1 ]
Epidemiologie
Laut Statistik gibt es nicht mehr als neun Fälle von Alkaptonurie pro 1 Million Einwohner. Und in den meisten europäischen Ländern gibt es einen Fall pro 100.000 bis 250.000 Lebendgeburten.
Eine Ausnahme unter den europäischen Ländern bildet die Slowakei (insbesondere die relativ kleine nordwestliche Region), wo die Prävalenz von Alkaptonurie bei einem Fall pro 19.000 Neugeborenen liegt. Dies ist höchstwahrscheinlich darauf zurückzuführen, dass unter den dort lebenden slowakischen Roma-Familien die Inzuchtrate (Ehen zwischen Cousins und Cousinen) mit 10-14 % die höchste in Europa ist. [ 2 ]
Ursachen Alkaptonurie
Die genauen Ursachen der Alkaptonurie als angeborene Störung des Katabolismus (Stoffwechselabbaus) der aromatischen (homozyklischen) α-Aminosäure Tyrosin sind geklärt: Diese Art von Stoffwechselstörung ist eine Folge homozygoter oder zusammengesetzter heterozygoter Mutationen eines der Tausenden von Genen auf Chromosom 3, genauer gesagt des HGD-Gens am Locus 3q21-q23 auf dem langen Arm des Chromosoms. Dieses Gen kodiert die Nukleotidsequenzen des Leberenzyms Homogentisat-1,2-Dioxygenase [ 3 ] (auch Homogentisinsäureoxidase oder Homogentisatoxidase genannt) - eines eisenhaltigen Metalloproteins, das für eine der Phasen des Tyrosinabbaus im Körper notwendig ist. [ 4 ], [ 5 ]
Somit handelt es sich bei der Alkaptonurie um einen Defekt des Enzyms Homogentisat-1,2-Dioxygenase, genauer gesagt um die Folge eines genetisch bedingten Mangels oder vollständigen Fehlens dieses Enzyms. [ 6 ]
Da es sich bei der Alkaptonurie um einen angeborenen Enzymmangel handelt, wird sie autosomal-rezessiv vererbt. Das bedeutet, dass beide Elternteile über ein verändertes Gen für das Enzym verfügen müssen, damit eine Alkaptonurie bei Kindern auftritt, da jeder von ihnen nur eine der beiden vorhandenen Kopien des Gens an das Kind weitergibt.
Den neuesten Daten zufolge gibt es mehr als zweihundert Modifikationsvarianten des HGD-Gens, wobei Missense-Mutationen, Translokation und Spleißen am häufigsten beobachtet werden.
Risikofaktoren
Der einzige Risikofaktor für die Entwicklung dieser angeborenen Enzymopathie ist das Vorhandensein einer solchen Erkrankung in der Familienanamnese und die Vererbung von zwei modifizierten Kopien des HGD-Gens, sofern die Eltern keine Alkaptonurie aufweisen (das Risiko der Übertragung der Anomalie beträgt 25 %) oder ein Elternteil an dieser Störung leidet. [ 7 ]
Pathogenese
Tyrosin spielt eine Schlüsselrolle bei der Proteinsynthese, der Produktion von Chromoproteinen – dem Hautpigment Melanin – sowie von Schilddrüsenhormonen und Katecholamin-Neurotransmittern.
Der Mechanismus zur Regulierung der Tyrosinmenge in Zellen ist sehr komplex, und der Körper normalisiert seinen überschüssigen Gehalt durch Abbau. Der Prozess des Tyrosinabbaus ist wie bei allen aromatischen Aminosäuren mehrstufig und verläuft in mehreren Phasen. Jede Phase des metabolischen Tyrosinabbaus erfolgt unter Beteiligung eines bestimmten Enzyms und der Bildung einer Zwischenverbindung.
Zunächst wird die Aminosäure zu Parahydroxyphenylpyruvat abgebaut, das in Alkapton – 2,5-Dihydroxyphenylessigsäure oder Homogentisinsäure – umgewandelt wird. Anschließend sollte das Alkapton in Maleinsäure umgewandelt werden, was jedoch nicht geschieht. [ 8 ]
Und die Pathogenese der Alkaptonurie besteht im Aufhören der biochemischen Reaktionen des Tyrosinkatabolismus im Stadium der Bildung von Homogentisinsäure: Es ist einfach kein Enzym erforderlich, um sie abzubauen – Homogentisatoxidase.
Homogentisinsäure wird vom Körper nicht verwendet und kann sich bei der Ausscheidung über die Nieren ansammeln. Darüber hinaus wird es zu Benzochinoacetat (Benzochinonessigsäure) oxidiert, das durch Bindung an die Moleküle von Geweben und Körperflüssigkeiten biopolymere Verbindungen bildet, die wie Melanin gefärbt sind.
Durch die Ansammlung dieser Zwischenprodukte im Gewebe kommt es zu einer Störung der Kollagenstruktur des Knorpelgewebes, wodurch dessen Elastizität abnimmt – mit dem Auftreten zahlreicher klinischer Anzeichen einer Alkaptonurie und der Entwicklung von Komplikationen.
Symptome Alkaptonurie
Alkaptonurie bei Neugeborenen und Säuglingen ist durch eine Verdunkelung des Urins gekennzeichnet. Bei Kontakt mit Luft verfärbt sich der Urin auf Windeln und Unterwäsche dunkelbraun. Dies ist auf die Ansammlung und Freisetzung von Homogentisinsäure zurückzuführen, die zu Benzochinoacetat oxidiert wird. [ 9 ]
Liegen keine weiteren Symptome vor, wird eine Alkaptonurie bei Kleinkindern oft nicht rechtzeitig erkannt, da sich der Urin nach mehreren Stunden des Wasserlassens verdunkeln kann. Einigen Daten zufolge wird nur ein Fünftel der Kinder unter 12 Monaten, die mit diesem Enzymmangel geboren wurden, klinisch diagnostiziert. Daher ist es für Eltern sehr wichtig, auf die Pflege ihrer Säuglinge zu achten.
Zu den frühen Anzeichen gehört außerdem eine Pigmentierung (bläulich-graue Farbe) der Lederhaut der Augen und der Knorpel der Ohren und der Nase, die oft als Ochronose bezeichnet wird.[ 10 ]
Mit der Zeit treten weitere Symptome auf:
- starke Pigmentierung der Haut an den Wangenknochen, Achselhöhlen und Genitalien;
- Fleckenbildung auf der Kleidung bei Kontakt mit verschwitzten Körperstellen;
- Anfälle allgemeiner Schwäche;
- heisere Stimme.
Es sollte berücksichtigt werden, dass Alkaptonurie und Ochronose, wie oben erwähnt, synonyme Bezeichnungen für dieselbe Störung des Tyrosinkatabolismus sind.
Ahornsirupkrankheit und Alkaptonurie. Die angeborene Ahornsirupkrankheit oder Leuzinose ist ebenfalls eine Stoffwechselstörung. Sie weist das gleiche Vererbungsmuster auf, und es treten sogar Mutationen auf demselben Chromosom auf. Sie betrifft jedoch das Gen, das den Enzymkomplex der verzweigtkettigen α-Ketosäure-Dehydrogenase kodiert. Dadurch kann der Körper bestimmte Proteinbestandteile, insbesondere die Aminosäuren Leucin, Isoleucin und Valin, nicht abbauen. Bei dieser Erkrankung riechen Urin (und Ohrenschmalz) süßlich. Darüber hinaus umfasst das klinische Bild dieser Art von organischer Azidämie Hypopigmentierung, Blutdruckschwankungen, Krampfanfälle, Erbrechen und Durchfall, einen Abfall des Blutzuckerspiegels, Ketoazidose, Halluzinationen usw. Die Sterblichkeitsrate bei Kindern ist recht hoch. Bei Erwachsenen kann es ohne Behandlung zu Koma und Tod aufgrund eines Hirnödems kommen.
Albinismus und Alkaptonurie werden nur durch Tyrosin „vereinigt“. Albinismus, einschließlich okulokutaner Albinismus, wird durch genetische Mutationen verursacht, die die Produktion des Pigments Melanin beeinträchtigen. Angeborene Veränderungen finden sich im TYR-Gen auf Chromosom 11 (11q14.3), das für Tyrosinase kodiert, ein kupferhaltiges Melanosomenzym, das für die Bildung von Hautpigmenten auf Basis von Tyrosinstoffwechselprodukten notwendig ist. Diese Erkrankung ist deutlich häufiger als Alkaptonurie.
Komplikationen und Konsequenzen
Die Folgen und Komplikationen der Alkaptonurie werden durch die Wirkung intermediärer Metaboliten von Tyrosin – Homogentisinsäure und Benzochinonessigsäure – verursacht und treten aufgrund der Ablagerung reaktiver pigmentierter Polymere, der Zerstörung von Kollagenfibrillen und der Verschlechterung des Zustands des Knorpels (mit einer Verringerung seiner Widerstandsfähigkeit gegen mechanische Beanspruchung) auf.
Im Erwachsenenalter entwickeln sich im Laufe der Jahre degenerative Arthritis und Osteoarthritis der großen Gelenke (Hüfte, Iliosakralgelenk und Knie); die Zwischenwirbelräume verengen sich (vor allem in der Lenden- und Brustwirbelsäule) – mit Verkalkung und Bildung von Osteophyten; die Gewebedichte der subchondralen Knochenplatten nimmt ab, und die darunter liegenden Knochen können einer pathologischen Umgestaltung mit Bildung von Wucherungen und Deformationen unterliegen. [ 11 ]
Aufgrund derselben Verkalkung können Schäden an den Herzklappen (Aorten- und Mitralklappen) und den Koronararterien beobachtet werden – mit Anzeichen einer koronaren Herzkrankheit sowie der Bildung von Steinen in den Nieren und der Prostata. [ 12 ], [ 13 ]
Diagnose Alkaptonurie
Typischerweise basiert die Diagnose angeborener Stoffwechselstörungen auf der Untersuchung biologischer Körperflüssigkeiten.
Welche Tests und Reaktionen können zur Diagnose einer Alkaptonurie beitragen? Urinuntersuchungen sind erforderlich, um Homogentisinsäure nachzuweisen und ihren Spiegel zu bestimmen (normal – 20–30 mg pro Tag, erhöht – 3–8 g). Eine Urinprobe wird mittels Gaschromatographie oder Massenspektrometrie untersucht. Mittels Flüssigkeitschromatographie ist ein Screening-Test auf Eisenchlorid im Urin möglich. [ 14 ]
Es gibt auch eine Methode zur Schnelldiagnostik – die Bestimmung von Alkapton in getrockneten Urinflecken auf Papier (anhand der Farbintensität).
Bei der Diagnoseklärung geht es bei der instrumentellen Diagnostik (Röntgenbild) darum, radiologische Anzeichen einer Arthrose und anderer Gelenkerkrankungen bei Patienten zu erkennen.
Die Diagnose wird durch molekulargenetische Methoden zur Diagnose von Erbkrankheiten, wie genetische Tests und DNA-Sequenzierung, bestätigt. [ 15 ]
Differenzialdiagnose
Zu den Differentialdiagnosen zählen Hämochromatose und akutes Leberversagen des Neugeborenen, Melaninurie, akute intermittierende Porphyrie, hämophagozytische Lymphohistiozytose, primäre mitochondriale Pathologie, rheumatoide Arthritis und ankylosierende Spondylitis.
Wen kann ich kontaktieren?
Behandlung Alkaptonurie
Die Hauptbehandlung der Alkaptonurie besteht in der oralen Gabe hoher Dosen (mindestens 1000 mg pro Tag) Ascorbinsäure. Bei Kindern erhöht dies die Ausscheidung von Homogentisinsäure im Urin, bei Erwachsenen reduziert es den Harngehalt ihres Derivats Benzochinonessigsäure und verlangsamt dessen Bindung an Bindegewebsstrukturen der Gelenke und an Kollagen. [ 16 ]
Westeuropäische Kliniken testen das Medikament Nitisinon (Orfalin), ein Medikament aus der Gruppe der Metaboliten, das die zweite Stufe des Tyrosinabbaus hemmt: die Umwandlung von para-Hydroxyphenylpyruvat in Homogentisinsäure. Die Anwendung dieses pharmakologischen Wirkstoffs führt jedoch zur Anreicherung von Tyrosin und kann schwere Nebenwirkungen wie Hornhauttrübung und Lichtscheu, Nasen- und Magenblutungen, Leberversagen, Blutveränderungen usw. verursachen. In den USA ist Nitisinon jedoch von der FDA zur Behandlung von Tyrosinämie Typ I zugelassen. [17 ], [ 18 ]
So werden bei Gelenkbeschwerden, die durch Alkaptonurie verursacht werden, physiotherapeutische Behandlungen durchgeführt – Bewegungstherapien zur Steigerung der Muskelkraft und Verbesserung der Gelenkbeweglichkeit, Balneotherapie und Peloidtherapie zur Schmerzlinderung.
Obwohl Tyrosin nicht nur über die Nahrung zugeführt, sondern auch im Körper produziert wird, wird Patienten mit Alkaptonurie empfohlen, eine eiweißarme Diät einzuhalten und den Verzehr von tyrosinreichen Lebensmitteln, vor allem Rind- und Schweinefleisch, Milchprodukten (vor allem Käse), Hülsenfrüchten, Nüssen und Samen, einzuschränken.
Verhütung
Eine Vorbeugung von Genmutationen ist nicht möglich. Um jedoch die Geburt von Kindern mit einem hohen Risiko für angeborene Erkrankungen zu verhindern, gibt es eine medizinisch-genetische Beratung, die vor einer geplanten Schwangerschaft für Paare notwendig ist, deren Familiengeschichte Erbkrankheiten umfasst. [ 19 ]
Prognose
Tödliche Folgen einer Alkaptonurie sind sehr selten. Schwere Komplikationen an Herz und Nieren können zum Tod führen. Daher ist die Lebenserwartung von Menschen mit Alkaptonurie insgesamt gut.
Allerdings ist die Lebensqualität aufgrund starker Schmerzen in den Gelenken oder der Wirbelsäule mit erheblicher, oft fortschreitender Einschränkung der Beweglichkeit eingeschränkt.