
Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
Martin-Bell-Syndrom
Facharzt des Artikels

Zuletzt überprüft: 04.07.2025
Das Martin-Bell-Syndrom wurde 1943 von Ärzten beschrieben und nach ihnen benannt. Es handelt sich um eine genetische Störung, die mit geistiger Behinderung einhergeht. 1969 wurden die für diese Krankheit charakteristischen Veränderungen des X-Chromosoms (Fragilität im distalen Arm) identifiziert. 1991 entdeckten Wissenschaftler das für die Entstehung dieser Krankheit verantwortliche Gen. Die Krankheit wird auch als „Fragiles-X-Syndrom“ bezeichnet. Sowohl Jungen als auch Mädchen sind anfällig für die Krankheit, Jungen sind jedoch dreimal häufiger betroffen.
Epidemiologie
Das Martin-Bell-Syndrom ist eine relativ häufige Erkrankung: 0,3 bis 1,0 von 1.000 Männern und 0,2 bis 0,6 von 1.000 Frauen leiden darunter. Darüber hinaus werden Kinder mit Martin-Bell-Syndrom auf allen Kontinenten gleich häufig geboren. Offensichtlich haben Nationalität, Hautfarbe, Augenform, Lebensbedingungen und Wohlbefinden der Menschen keinen Einfluss auf das Auftreten der Krankheit. Ihre Häufigkeit ist nur mit der Häufigkeit des Down-Syndroms vergleichbar (1 Erkrankung von 600 bis 800 Neugeborenen). Ein Fünftel der männlichen Träger des veränderten Gens ist gesund und weist keine klinischen oder genetischen Anomalien auf. Der Rest hat Anzeichen einer geistigen Behinderung von leichter bis schwerer Form. Unter den weiblichen Trägern ist etwas mehr als ein Drittel erkrankt.
Das Fragile-X-Syndrom betrifft etwa 1 von 2.500–4.000 Männern und 1 von 7.000–8.000 Frauen. Die Prävalenz der Trägerschaft bei Frauen wird auf 1 von 130–250 geschätzt; bei Männern liegt sie bei 1 von 250–800.
Ursachen Martin-Bell-Syndrom
Das Martin-Bell-Syndrom entsteht durch den vollständigen oder teilweisen Produktionsstopp eines bestimmten Proteins im Körper. Dies ist auf die fehlende Reaktion des auf dem X-Chromosom lokalisierten FMR1-Gens zurückzuführen. Die Mutation entsteht durch die Umstrukturierung des Gens aus instabilen Strukturvarianten der Genzustände (Allele) und nicht von Anfang an. Die Krankheit wird ausschließlich in der männlichen Linie übertragen, und der Mann muss nicht zwangsläufig erkrankt sein. Männliche Träger geben das Gen unverändert an ihre Töchter weiter, sodass deren geistige Behinderung nicht offensichtlich ist. Bei weiterer Weitergabe des Gens von der Mutter an ihre Kinder mutiert das Gen, und alle für diese Krankheit charakteristischen Symptome treten auf.
Pathogenese
Die Pathogenese des Martin-Bell-Syndroms beruht auf Mutationen des Genapparats, die die Produktion des FMR-Proteins blockieren. Dieses Protein ist für den Körper, insbesondere in Neuronen, lebenswichtig und kommt in verschiedenen Geweben vor. Untersuchungen zeigen, dass FMR-Proteine direkt an der Regulation der Translation im Hirngewebe beteiligt sind. Das Fehlen dieses Proteins oder seine eingeschränkte Produktion durch den Körper führt zu geistiger Behinderung.
Bei der Pathogenese der Erkrankung gilt die Genhypermethylierung als Schlüsselstörung, allerdings ist es bisher nicht gelungen, den Entstehungsmechanismus dieser Störung eindeutig zu identifizieren.
Gleichzeitig wurde auch eine Locus-Heterogenität der Pathologie entdeckt, die sowohl mit Polyallelismus als auch mit Polylocus assoziiert ist. Es wurde das Vorhandensein allelischer Varianten der Krankheitsentwicklung festgestellt, die durch das Vorhandensein von Punktmutationen sowie die Zerstörung des FMRL-Typ-Gens verursacht werden.
Die Patienten haben außerdem zwei fragile, folsäureempfindliche Triplets, die 300 kb sowie 1,5–2 Millionen bp vom fragilen Triplet entfernt liegen, das das FMR1-Gen enthält. Der Mechanismus der in den Genen FRAXE und FRAXF auftretenden Mutationen (sie werden in den oben genannten fragilen Triplets identifiziert) hängt mit dem Störungsmechanismus des Martin-Bell-Syndroms zusammen. Dieser Mechanismus wird durch die Ausbreitung von GCC- und CGG-Wiederholungen verursacht, die eine Methylierung der sogenannten CpG-Inseln verursachen. Neben der klassischen Form der Pathologie gibt es auch zwei seltene Typen, die sich durch die Ausbreitung von Trinukleotid-Wiederholungen unterscheiden (bei männlicher und weiblicher Meiose).
Es wurde festgestellt, dass dem Patienten bei der klassischen Form des Syndroms ein spezielles nukleozytoplasmatisches Protein vom Typ FMR1 fehlt, das die Funktion der Bindung verschiedener mRNAs übernimmt. Darüber hinaus fördert dieses Protein die Bildung eines Komplexes, der Translationsprozesse innerhalb der Ribosomen unterstützt.
Symptome Martin-Bell-Syndrom
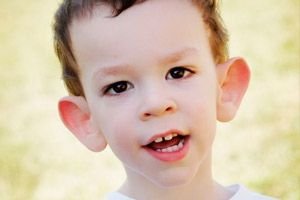
Wie erkennt man die Krankheit bei Kindern? Was sind die ersten Anzeichen? In den ersten Lebensmonaten eines Kindes ist das Martin-Bell-Symptom nicht zu erkennen, außer dass manchmal eine Abnahme des Muskeltonus beobachtet wird. Nach einem Jahr ist das klinische Bild der Krankheit deutlicher: Das Kind beginnt spät zu laufen und zu sprechen, manchmal fehlt die Sprache vollständig. Es ist hyperaktiv, fuchtelt willkürlich mit den Armen, hat Angst vor Menschenmengen und Lärm, ist stur, hat heftige Wutausbrüche, ist emotional instabil, hat epileptische Anfälle und vermeidet Augenkontakt. Bei Patienten mit Martin-Bell-Syndrom verrät die Krankheit auch ihr Aussehen: Die Ohren sind abstehend und groß, die Stirn ist schwer, das Gesicht ist länglich, das Kinn steht hervor, es treten Strabismus, breite Hände und Füße auf. Sie sind auch durch endokrine Störungen gekennzeichnet: oft hohes Gewicht, Fettleibigkeit, große Hoden bei Männern, frühe Pubertät.

Bei Patienten mit Martin-Bell-Syndrom variiert der Intelligenzgrad stark: von leichter geistiger Behinderung bis hin zu schweren Fällen. Wenn ein normaler Mensch im Durchschnitt einen Intelligenzquotienten (IQ) von 100 und ein Genie von 130 hat, liegt der IQ bei anfälligen Menschen bei 35-70.
Alle klinischen Symptome der Pathologie können durch eine Trias von Hauptmanifestationen charakterisiert werden:
- Oligophrenie (IQ liegt bei 35–50);
- Dysmorphophobie (es werden abstehende Ohren und Prognathie beobachtet);
- Makroorchismus, der nach Beginn der Pubertät auftritt.
Bei etwa 80 % der Patienten liegt zudem ein Bikuspidalklappenprolaps vor.
Die Vollform des Syndroms manifestiert sich jedoch nur bei 60 % aller Patienten. Bei 10 % wird nur eine geistige Behinderung festgestellt, bei den übrigen entwickelt sich die Krankheit mit einer anderen Kombination von Symptomen.
Zu den ersten Anzeichen der Krankheit, die bereits in jungen Jahren auftreten, gehören:
- das kranke Kind weist im Vergleich zur Entwicklung anderer Gleichaltriger eine erhebliche geistige Behinderung auf;
- Aufmerksamkeits- und Konzentrationsstörungen;
- starke Sturheit;
- Kinder beginnen erst sehr spät zu laufen und zu sprechen;
- Es werden Hyperaktivität und Sprachentwicklungsstörungen beobachtet;
- sehr starke und unkontrollierbare Wutanfälle;
- Es kann zu Mutismus kommen, d. h. zu einem völligen Sprachverlust bei einem Kind.
- das Baby leidet unter sozialer Angst und kann aufgrund von Lärm oder anderen lauten Geräuschen in Panik geraten;
- das Kind fuchtelt unkontrolliert und chaotisch mit den Armen;
- es wird Schüchternheit beobachtet, das Kind hat Angst, sich an überfüllten Orten aufzuhalten;
- das Auftreten verschiedener Zwangsgedanken, instabiler emotionaler Zustand;
- Das Baby scheut sich möglicherweise davor, Menschen in die Augen zu sehen.
Bei Erwachsenen werden folgende Symptome der Pathologie beobachtet:
- spezifisches Erscheinungsbild: ein längliches Gesicht mit schwerer Stirn, großen abstehenden Ohren, einem stark vorstehenden Kinn;
- Plattfüße, Mittelohrentzündung und Strabismus;
- die Pubertät setzt recht früh ein;
- es kann zu Fettleibigkeit kommen;
- Ziemlich häufig werden beim Martin-Bell-Syndrom Herzfehler beobachtet;
- bei Männern wird eine Vergrößerung der Hoden beobachtet;
- die Gelenkverbindungen werden sehr beweglich;
- Gewicht und Größe nehmen stark zu.
Diagnose Martin-Bell-Syndrom
Um das Martin-Bell-Syndrom zu diagnostizieren, müssen Sie sich an einen qualifizierten Genetiker wenden. Die Diagnose wird nach spezifischen genetischen Tests gestellt, die es ermöglichen, das defekte Chromosom zu identifizieren.
Tests
In einem frühen Stadium der Erkrankung wird eine zytogenetische Methode angewendet, bei der dem Patienten ein Fragment von Zellmaterial entnommen und anschließend Folsäure zugesetzt wird, um Veränderungen der Chromosomen hervorzurufen. Nach einer gewissen Zeit wird ein Bereich des Chromosoms identifiziert, in dem eine auffällige Ausdünnung vorliegt – dies ist ein Zeichen für das Vorliegen eines Fragilen-X-Syndroms.
Für die Diagnose im Spätstadium der Erkrankung ist dieser Test allerdings nicht geeignet, da seine Aussagekraft durch die weitverbreitete Einnahme folsäurehaltiger Multivitaminpräparate eingeschränkt ist.
Die integrierte Diagnostik des Martin-Bell-Syndroms ist eine molekulargenetische Untersuchung, die darin besteht, die Anzahl der sogenannten Trinukleotid-Repeats im Gen zu bestimmen.
 [ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
Instrumentelle Diagnostik
Eine hochspezifische Methode der instrumentellen Diagnostik ist die PCR (Polymerase-Kettenreaktion), die es ermöglicht, die Struktur der im X-Chromosom enthaltenen Aminosäurereste zu untersuchen und so das Vorliegen des Martin-Bell-Syndroms festzustellen.
Es gibt auch eine separate, noch spezifischere Methode zur Pathologiediagnostik – eine Kombination aus PCR und Nachweis mittels Kapillarelektrophorese. Diese Methode ist hochpräzise und erkennt Chromosomenpathologien bei Patientinnen mit primärer Ovarialinsuffizienz sowie Ataxie-Syndrom.
Das Vorhandensein des Defekts kann nach der Durchführung einer EEG-Diagnostik festgestellt werden. Patienten mit dieser Krankheit haben eine ähnliche bioelektrische Gehirnaktivität.
Welche Tests werden benötigt?
Differenzialdiagnose
Zu den differenzierten Methoden, die bei der Verdachtsdiagnose helfen, zählen:
- klinisch – 97,5 % der Patienten weisen deutliche Anzeichen einer geistigen Behinderung (mittelschwer oder schwer) auf; 62 % haben abstehende große Ohren; 68,4 % haben ein großes, vorstehendes Kinn und eine große, vorstehende Stirn; 68,4 % der Jungen haben vergrößerte Hoden, 41,4 % weisen Sprachbesonderheiten auf (ungleichmäßige Sprechgeschwindigkeit, unkontrollierbare Lautstärke usw.);
- zytogen - Blut wird auf Lymphozytenkultur untersucht, die Anzahl der Zellen mit fragilem X-Chromosom pro 100 untersuchten Zellen wird bestimmt;
- Elektroenzephalographie – Dabei werden Veränderungen der elektrischen Impulse des Gehirns aufgezeichnet, die spezifisch für das Martin-Bell-Syndrom sind.
Wen kann ich kontaktieren?
Behandlung Martin-Bell-Syndrom
Bei der Behandlung erwachsener Patienten werden Antidepressiva mit Psychostimulanzien eingesetzt. Der Verlauf der medikamentösen Therapie wird ständig von einem Psychologen und Psychiater überwacht. Darüber hinaus werden in Privatkliniken Mikroinjektionen mit Medikamenten wie Cerebrolysin (oder seinen Derivaten) sowie Cytomedinen (wie Solcoseryl oder Lidase) durchgeführt.
Bei der Entwicklung eines ataktischen Syndroms werden blutverdünnende Medikamente und Nootropika eingesetzt. Zusätzlich werden Aminosäuremischungen und Angioprotektoren verschrieben. Frauen mit primärem Ovarialversagen wird eine Korrekturbehandlung mit pflanzlichen Arzneimitteln und Östrogenen verschrieben.
Auch Glutaminrezeptor-Antagonisten werden zur Behandlung eingesetzt.
Traditionell umfasst die Behandlung des Martin-Bell-Syndroms die Einnahme von Medikamenten, die die Symptome der Krankheit, aber nicht ihre Ursache, beeinflussen. Diese Therapie beinhaltet die Verschreibung von Antidepressiva, Neuroleptika und Psychostimulanzien. Nicht alle Medikamente sind für die Anwendung bei Kindern geeignet, daher ist die Liste der Medikamente recht begrenzt. Zu den Neuroleptika, die nach 3 Jahren (dem frühesten Verschreibungsalter) angewendet werden können, gehören Haloperidol in Tropfen und Tabletten, Chlorpromazinlösung und Periciazin in Tropfen. Daher wird die Haloperidol-Dosis für Kinder in Abhängigkeit vom Körpergewicht berechnet. Für Erwachsene wird die Dosis individuell verordnet. Die orale Einnahme erfolgt, beginnend mit 0,5–5 mg 2–3-mal täglich, dann wird die Dosis schrittweise auf 10–15 mg erhöht. Bei Besserung wird auf eine niedrigere Dosis umgestellt, um den erreichten Zustand zu halten. Bei psychomotorischer Unruhe werden 5–10 mg intramuskulär oder intravenös verabreicht; mehrere Wiederholungen sind nach 30–40 Minuten möglich. Die Tagesdosis sollte 100 mg nicht überschreiten. Nebenwirkungen wie Übelkeit, Erbrechen, Muskelkrämpfe, erhöhter Blutdruck, Herzrhythmusstörungen usw. sind möglich. Ältere Menschen sollten besondere Vorsichtsmaßnahmen treffen, da Fälle von plötzlichem Herzstillstand registriert wurden und Spätdyskinesien (unwillkürliche Bewegungen) auftreten können.
Antidepressiva steigern die Aktivität der Gehirnstrukturen, lindern Depressionen, Verspannungen und verbessern die Stimmung. Zu diesen Medikamenten, die für das Martin-Bell-Syndrom im Alter von 5 bis 8 Jahren empfohlen werden, gehören Clomipromin, Sertralin, Fluoxetin und Fluvoxamin. Fluoxetin wird 1-2-mal (vorzugsweise in der ersten Tageshälfte) oral zu den Mahlzeiten eingenommen, beginnend mit 20 mg pro Tag und bei Bedarf auf 80 mg erhöht. Älteren Menschen wird eine Dosis von mehr als 60 mg nicht empfohlen. Der Behandlungsverlauf wird vom Arzt festgelegt, beträgt jedoch nicht mehr als 5 Wochen.
Mögliche Nebenwirkungen: Schwindel, Angstzustände, Tinnitus, Appetitlosigkeit, Tachykardie, Ödeme usw. Vorsicht ist geboten bei der Verschreibung an ältere Menschen, Menschen mit Herz-Kreislauf-Erkrankungen und Diabetes.
Psychostimulanzien sind Psychopharmaka, die zur Steigerung der Wahrnehmung äußerer Reize eingesetzt werden: Sie schärfen das Gehör, die Reaktionsbereitschaft und das Sehvermögen.
Diazepam wird als Beruhigungsmittel bei Neurosen, Angstzuständen, epileptischen Anfällen und Krämpfen verschrieben. Es wird oral, intravenös, intramuskulär und rektal (in den Enddarm) eingenommen. Es wird individuell, abhängig vom Schweregrad der Erkrankung, mit den kleinsten Dosen von 5-10 mg, täglich - 5-20 mg verschrieben. Die Behandlungsdauer beträgt 2-3 Monate. Bei Kindern wird die Dosis unter Berücksichtigung des Körpergewichts und der individuellen Merkmale berechnet. Nebenwirkungen sind Lethargie, Apathie, Schläfrigkeit, Übelkeit und Verstopfung. Die Kombination mit Alkohol ist gefährlich, eine Abhängigkeit von der Droge ist möglich.
Bei der Behandlung des Martin-Bell-Syndroms kam es mit der Einführung von Medikamenten aus tierischem Material (Gehirn) zu einer Besserung des Zustands: Cerebrolysat, Cerebrolysin, Cerebrolysat-M. Die Hauptbestandteile dieser Medikamente sind Peptide, die die Proteinproduktion in Neuronen fördern und so den Proteinmangel kompensieren. Cerebrolysin wird als 5-10 ml-Strahl verabreicht, die Behandlung umfasst 20-30 Injektionen. Das Medikament wird Kindern ab einem Jahr verschrieben und einen Monat lang täglich 1-2 ml intramuskulär verabreicht. Wiederholte Verabreichungen sind möglich. Nebenwirkungen in Form von Fieber, kontraindiziert für Schwangere.
Es gab Versuche, die Krankheit mit Folsäure zu behandeln, aber nur der Verhaltensaspekt verbesserte sich (Aggression und Hyperaktivität nahmen ab, die Sprache verbesserte sich), und auf intellektueller Ebene änderte sich nichts. Um den Krankheitszustand zu verbessern, wird Folsäure verschrieben, physiotherapeutische Methoden, Logopädie, pädagogische und soziale Korrektur sind angezeigt.
Lithiumpräparate gelten ebenfalls als wirksam, da sie die Anpassung des Patienten an das soziale Umfeld sowie die kognitive Aktivität verbessern. Darüber hinaus regulieren sie auch sein Verhalten in der Gesellschaft.
Die Anwendung von Kräutern beim Martin-Bell-Syndrom ist als Antidepressivum möglich. Zu den Kräutern, die Verspannungen und Angstzustände lindern und den Schlaf verbessern, gehören Baldrian, Pfefferminze, Thymian, Johanniskraut und Kamille. Aufgüsse werden wie folgt zubereitet: Für 1 Teelöffel trockene Kräuter benötigen Sie ein Glas kochendes Wasser. Die Abkochungen werden mindestens 20 Minuten lang aufgegossen und hauptsächlich abends vor dem Schlafengehen oder nachmittags eingenommen. Ein Löffel Honig wäre eine gute Ergänzung.
Physiotherapeutische Behandlung
Um neurologische Manifestationen zu beseitigen, werden spezielle physiotherapeutische Verfahren durchgeführt – beispielsweise Übungen im Pool, Muskelentspannung und Akupunktur.
Chirurgische Behandlung
Ein wichtiger Schritt in der Behandlung sind auch plastisch-chirurgische Eingriffe – Operationen, die das Aussehen des Patienten verbessern. Plastische Operationen werden an den Gliedmaßen und Ohrmuscheln sowie an den Genitalien durchgeführt. Auch die Korrektur von Gynäkomastie mit Epispadie sowie anderer Schönheitsfehler wird durchgeführt.
Verhütung
Die einzige Möglichkeit, der Krankheit vorzubeugen, ist die pränatale Untersuchung schwangerer Frauen. Es gibt spezielle Untersuchungen, die eine frühzeitige Erkennung der Pathologie ermöglichen. Danach wird ein Schwangerschaftsabbruch empfohlen. Alternativ wird eine IVF eingesetzt, die dem Kind helfen kann, ein gesundes X-Chromosom zu erben.
Die Prävention des Patienten hängt davon ab, ob die Genmutation erneut aufgetreten ist oder vererbt wurde. Hierzu wird eine molekulargenetische Diagnostik durchgeführt. Die Tatsache, dass der Test bei Verwandten kein „fragiles X-Chromosom“ zeigte, spricht für die „Frische“ der Mutation, was bedeutet, dass das Risiko, ein Kind mit Martin-Bell-Syndrom zu bekommen, sehr gering ist. In Familien mit erkrankten Personen hilft der Test, Wiederholungsfälle zu vermeiden.
Prognose
Die Prognose des Martin-Bell-Syndroms ist lebenslang günstig, die Genesungschancen jedoch schlecht. Die Lebenserwartung hängt vom Schweregrad der Erkrankung und den damit verbundenen Defekten ab. Der Patient kann ein normales Leben führen. Bei schweren Formen des Martin-Bell-Syndroms besteht das Risiko einer lebenslangen Behinderung.
Lebenserwartung
Das Martin-Bell-Syndrom hat keine schwerwiegenden negativen Auswirkungen auf die Gesundheit, daher unterscheidet sich die Lebenserwartung der meisten Menschen, bei denen diese Krankheit diagnostiziert wurde, nicht von den Standardindikatoren.