
Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
Holoprosencephalie beim Fötus und Neugeborenen: Ursachen und Prognose
Facharzt des Artikels

Zuletzt überprüft: 04.07.2025
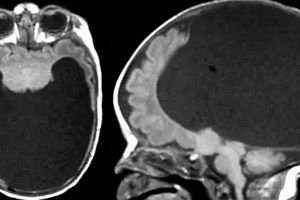
Bei der Holoprosenzephalie handelt es sich um eine relativ seltene Erkrankung, eine Störung der intrauterinen Entwicklung, bei der das Septum in der vorderen Hirnblase fehlt oder unterentwickelt ist. Das heißt, das Vorderhirn weist keine klare Unterteilung in zwei Hemisphären auf.
Epidemiologie
Holoprosenzephalie gilt nicht als sehr häufiger Defekt: Laut Statistik tritt die Krankheit bei 0,06–0,2 Neugeborenen pro 1.000 auf. Bei einem spontanen Schwangerschaftsabbruch können bis zu 4 pathologische Fälle pro 1.000 beobachtet werden.
In der Liste der Entwicklungsstörungen des zentralen Nervensystems wird Holoprosenzephalie in 2-3 % der Fälle sowie bei etwa 5 % der Föten während der Autopsie (Nekropsie) diagnostiziert.
Bei mehr als der Hälfte der Patienten ist die Holoprosenzephalie isoliert, in etwa 22 % der Fälle tritt sie in Kombination mit einer Vielzahl anderer Entwicklungsstörungen auf. In mehr als 17 % der Fälle wird eine syndromale Variante der Erkrankung diagnostiziert.
Bei Mädchen wird häufiger eine Holoprosenzephalie diagnostiziert.
Ursachen Holoprosencephalie
Holoprosenzephalie ist eine angeborene Entwicklungsstörung, die durch eine Störung des intrauterinen Wachstumsmechanismus des Fötus entsteht. Die Störung wird üblicherweise autosomal-rezessiv, autosomal-dominant und X-chromosomal vererbt.
Eine der Ursachen, die nicht mit Chromosomenanomalien zusammenhängt, ist eine genetische Mutation. Derzeit wurden folgende Gene identifiziert, die anfällig für Mutationen mit anschließender Entwicklung einer Holoprosenzephalie sind:
- SIX3 2p21;
- TGIF 18p11.3;
- ZIC2 13q32;
- SHH 7q36.
Experten weisen auch auf die negativen Auswirkungen endokriner Erkrankungen der Mutter, des Rauchens, des Alkoholkonsums und der Einnahme von Medikamenten aus der Salicylat-Reihe hin.
Risikofaktoren
Die Faktoren, die zur Entwicklung der Holoprosenzephalie beitragen, werden üblicherweise in erbliche und externe Faktoren unterteilt.
Eine Reihe erblicher Faktoren sind Chromosomenstörungen, die zu Veränderungen des Karyotyps führen:
- Orbeli-Syndrom;
- Ecardi-Syndrom;
- Meckel-Syndrom;
- Trisomie 13.
Äußere Faktoren der Holoprosenzephalie sind:
- endokrine Erkrankungen der werdenden Mutter, insulinabhängiger Diabetes mellitus;
- niedriger Cholesterinspiegel;
- Konsum alkoholischer Getränke;
- Einnahme bestimmter Medikamente (Salicylate, Methotrexat, Retinsäure, Misoprostol, Diphenylhydantoin).
Pathogenese
Die nicht-syndromale hereditäre Holoprosenzephalie wird am häufigsten autosomal-dominant vererbt, aber auch andere Vererbungswege sind möglich. Etwa 30–50 % der Erkrankungen weisen einen eindeutigen Zusammenhang mit einer Chromosomenstörung auf: Eine vorgegebene Verteilung der Defekte weist auf das Vorhandensein von mindestens zwölf verschiedenen Loci hin.
Das erste entdeckte mutierte Gen ist SHH, das sich am Locus 7q36 befindet. Mutationsveränderungen in SHH verursachen etwa 35 % der familiären Anomalien mit autosomal-dominanter Holoprosenzephalie.
Der Eiweißstoff SHH ist ein Signalprotein, das sowohl bei Tieren als auch bei Arthropoden produziert wird und für die normale fetale Entwicklung notwendig ist.
Mutationsbedingte Veränderungen dieser Proteinsubstanz führen zu Funktionsstörungen – Holoprosenzephalie. So gehören bestimmte zytogenetische Defekte, die den Prozess der Übertragung genetischer Informationen von DNA über RNA auf Proteine und Polypeptide beeinflussen, zu Translokationen, die sich auf 15–256 Kilobasen am 5'-Ende der codierten Episode des SHH-Gens erstrecken. Diese Arten von Chromosomenmutationen werden als positionell bezeichnet, da sie keine Veränderung der codierenden Sequenz bewirken, sondern die Kontrolle stören und die Struktur des Chromatins verändern, was den Prozess der Übertragung genetischer Informationen SHH beeinflusst.
Symptome Holoprosencephalie
Moderne Spezialisten unterscheiden verschiedene Arten der Holoprosenzephalie, die sich in Symptomen, Schweregrad und Häufigkeit unterscheiden. Genetiker können den genauen Grund für das Auftreten der Krankheit in der einen oder anderen Form noch immer nicht genau bestimmen. Vermutlich wird die Entwicklung einer bestimmten Form der Holoprosenzephalie durch den Ort der Chromosomenschädigung, die Merkmale der Schwangerschaft oder andere Faktoren beeinflusst.
Die Medizin kennt folgende Formen der Holoprosenzephalie:
- Die Alobare Holoprosenzephalie ist die komplexeste Form dieser Erkrankung. Sie weist schwere Entwicklungsstörungen auf, die das Gehirn, den Gesichtsbereich und andere Organe betreffen. 20 % der Fälle dieser Erkrankung sind von dieser Form betroffen.
- Die semilobäre Holoprosenzephalie ist die häufigste Form der Erkrankung und weist komplexe, aber weniger ausgeprägte Entwicklungsstörungen auf. Bei etwa 50 % aller Patienten wird der semilobäre Typ diagnostiziert.
- Die lobäre Holoprosenzephalie ist die mildeste Form der Erkrankung und lässt sich relativ gut chirurgisch und medikamentös korrigieren. Diese Erkrankung tritt bei etwa 20 % der Patienten mit Holoprosenzephalie auf.
Es gibt auch eine vierte Art der Holoprosenzephalie, die erst vor etwas mehr als zwanzig Jahren getrennt von anderen Arten betrachtet wurde. Dies ist eine seltene Form der mittelhemisphärischen Fusion - sie ist durch verschwommene Anzeichen gekennzeichnet, die sich vom klassischen Krankheitsverlauf unterscheiden.
 [ 21 ], [ 22 ], [ 23 ], [ 24 ], [ 25 ], [ 26 ], [ 27 ]
[ 21 ], [ 22 ], [ 23 ], [ 24 ], [ 25 ], [ 26 ], [ 27 ]
Erste Anzeichen
Die Symptome der Holoprosenzephalie können je nach Art der Erkrankung sehr unterschiedlich sein. Nur wenige frühe Anzeichen können häufig sein:
- Gaumenspalte und Lippenspalte;
- periodische Krampfanfälle;
- geistige Unzulänglichkeit;
- beeinträchtigte Reflexe;
- krankhafte Veränderungen der Hornhaut und Netzhaut.
- Der alobäre Typ der Holoprosenzephalie ist durch Zyklopenismus, Unterentwicklung der Nase, erhebliche Größenunterschiede im Kopf und multiple Defekte anderer Organe gekennzeichnet. Die Entwicklung des alobären Typs endet in den allermeisten Fällen mit einem spontanen Schwangerschaftsabbruch (oder einer Totgeburt): Die wenigen überlebenden Babys sterben innerhalb der ersten sechs Lebensmonate.
- Der semilobäre Typ der Holoprosenzephalie weist ebenfalls charakteristische Symptome auf: eine enge Anordnung der Augenhöhlen, eine leichte Abnahme des Kopfes und eine beeinträchtigte Entwicklung der Nasengänge. Säuglinge mit solchen Defekten sterben in den ersten 24 Lebensmonaten.
- Der Lappentyp ist durch Anomalien in der Struktur des Gaumens und der Oberlippe gekennzeichnet. Kinder mit einer solchen Pathologie können ein höheres Alter erreichen, sofern rechtzeitig ein chirurgischer Eingriff durchgeführt wird.
- Bei einer durchschnittlichen interhemisphärischen Fusion treten im Gesicht des Kindes keine Defekte auf. Es kommt jedoch zu geistiger Behinderung, Krampfanfällen und anderen neurologischen Symptomen.
In den meisten Fällen wird eine fetale Holoprosenzephalie während der Schwangerschaft oder unmittelbar nach der Geburt des Kindes festgestellt. Neben äußeren Manifestationen zeigen sich bei der Patientin endokrine Dysfunktionen, Nierendysplasie, Schäden an den Atemwegen und anderen Organen. Häufig werden Herzfehler, Autoimmunerkrankungen, fehlende Reflexe usw. diagnostiziert.
Komplikationen und Konsequenzen
Die Folgen einer Holoprosenzephalie hängen grundsätzlich vom Ausmaß der Hirnschädigung ab.
Bei der alobären Holoprosenzephalie ist ein tödlicher Ausgang am wahrscheinlichsten.
In anderen Fällen leiden die Kinder nicht nur an geistiger Behinderung, sondern auch an Krampfanfällen, Reflexproblemen und Funktionsstörungen des Hirnstamms.
Laut Statistik überleben mehr als 40 % der Kinder mit Holoprosenzephalie nicht länger als 5–6 Monate, etwa 80 % sterben im ersten Lebensjahr.
Beim lobären Typ der Erkrankung kann ein Kind zwar mehrere Jahre überleben, stirbt aber dennoch an dysfunktionalen Mangelerscheinungen.
Diagnose Holoprosencephalie
Jeder Kinderarzt oder Neonatologe kann ein gesundes Kind leicht von einem Baby mit Holoprosenzephalie unterscheiden, da die Manifestationen der Krankheit sehr spezifisch sind.
Die Diagnose einer Holoprosenzephalie erfolgt häufig im ersten Trimenon der Schwangerschaft anhand der Ergebnisse einer Ultraschalluntersuchung.
Holoprosenzephalie im Ultraschall manifestiert sich durch eine stark abnorme Struktur des Schädels und des Gehirns des ungeborenen Kindes:
- Beim alobären Typ der Holoprosenzephalie hat das Gehirn das Aussehen einer Blase mit flüssigem Inhalt, ohne die geringsten Anzeichen unterschiedlicher Hemisphären.
- Beim semilobären Typ der Erkrankung zeigt die Ultraschalluntersuchung eine besondere Furche, die den hinteren Teil des Gehirns durchzieht und einer teilweisen Abgrenzung in Hemisphären entspricht.
- Beim lobären Typ der Holoprosenzephalie ist die Diagnose etwas schwierig: Symptome einer unvollständigen Abgrenzung des Gehirns werden nur in den Tiefen des Organs festgestellt, da überwiegend Thalamus, Corpus callosum und Ventrikel betroffen sind.
Um eine Zuckerkrankheit (Diabetes) bei der werdenden Mutter festzustellen, werden bei Frauen Blut- und Urinuntersuchungen durchgeführt. Dadurch lässt sich eine Entwicklungsstörung des Fötus vermuten.
Die instrumentelle Diagnostik der Holoprosenzephalie in der pränatalen Phase erfolgt mittels molekulargenetischer Methoden. Untersuchungsmaterial wird bei Verdacht auf angeborene Anomalien entnommen, wenn die Indikation für eine Ultraschalluntersuchung unzureichend ist, wenn in der Vergangenheit Kinder mit verschiedenen angeborenen Pathologien geboren wurden, wenn die Eltern bestimmte Anzeichen für genetische Probleme aufweisen oder wenn die werdende Mutter an Diabetes mellitus leidet. Zur Materialentnahme werden Amniozentese oder Chorionzottenbiopsie verwendet. Genetische Tests bestehen oft aus der direkten Trennung des SHH-Gens zur Feststellung von Störungen oder der Untersuchung des Karyotyps des ungeborenen Kindes zur Beurteilung chromosomaler Inkonsistenzen. Es ist erwähnenswert, dass der Test in mehr als der Hälfte der Fälle keinen Karyotypfehler feststellt, weshalb diese Methode als nicht aussagekräftig gilt.
Was muss untersucht werden?
Welche Tests werden benötigt?
Differenzialdiagnose
Die Differentialdiagnostik der lobären Holoprosenzephalie erfolgt mit der septo-optischen Dysplasie. Wichtige Unterscheidungsmerkmale sind in diesem Fall:
- hypoplastische Veränderungen in den Frontallappen;
- Fehlbildung der Schläfenhörner in den Seitenventrikeln;
- Fehlen von Stirnhörnern.
Im pränatalen Stadium sollte die Holoprosenzephalie von einer schweren Ventrikulomegalie, einer Enzephalozele, zerebralen zystischen Formationen und einer Hydranenzephalie unterschieden werden.
Wen kann ich kontaktieren?
Behandlung Holoprosencephalie
Die Holoprosenzephalie erfordert keine spezielle Behandlung. Wenn der Arzt es für angebracht hält, kann er eine chirurgische Behandlung mit Korrektur der Gesichtsdefekte verschreiben. Eine symptomatische Behandlung wird ebenfalls durchgeführt, um die Beschwerden des Kindes zu lindern.
Bei den alobären und semilobären Formen der Holoprosenzephalie sind neurochirurgische Eingriffe nicht möglich, da der Zustand des Babys möglicherweise nicht nur nicht korrigiert wird, sondern sich auch verschlimmern kann.
Der lobäre Typ der Erkrankung kann chirurgisch behandelt werden: Defekte am Obergaumen und an der Lippe werden umgehend beseitigt und ausreichende Nasengänge gebildet.
Alle Formen der Pathologie erfordern den Einsatz einer antikonvulsiven Therapie, mit möglicher individueller Korrektur anderer Störungen.
Der Arzt kann bei Holoprosenzephalie folgende Medikamente verschreiben:
Dosierung und Art der Anwendung |
Nebenwirkungen |
Die besonderen Hinweise |
|
Suxilep |
Die Dosierung wird individuell verschrieben, etwa 5 mg pro kg Körpergewicht. |
Dyskinesie, Schwindel, Schwäche. |
Das Arzneimittel kann eine myelotoxische Wirkung haben. |
Sibazon |
Wird in individuellen Dosierungen verschrieben. |
Muskelschwäche, Schluckauf, Schlafstörungen. |
Bei längerem Gebrauch kann eine Arzneimittelabhängigkeit entstehen. |
Mydocalm |
Verschrieben in einer Dosierung von 5 mg pro kg Körpergewicht, dreimal täglich. |
Muskelschwäche, niedriger Blutdruck. |
Das Medikament wird Kindern unter 3 Jahren nicht verschrieben. |
Cerebrolysin |
Wird in individuellen Dosierungen verschrieben. |
Dyspepsie, Zittern, Hitzegefühl, Allergie. |
Die Injektionen des Arzneimittels werden langsam durchgeführt, um lokale Nebenwirkungen zu vermeiden. |
Vitamine
Vitamine sind für die Linderung des Zustands eines Kindes mit Holoprosenzephalie nicht von entscheidender Bedeutung. In einigen Fällen kann jedoch eine Vitamintherapie in Erhaltungsdosen in Betracht gezogen werden:
Art des Vitamins |
Wirkung von Vitamin |
Dosierung |
Vitamin A |
Verbessert das Sehvermögen und den Zustand der Schleimhäute. |
1250 IE |
Vitamin D |
Sorgt für den Kalzium- und Phosphorstoffwechsel und verbessert die Knochengewebebildung. |
300 IE |
Ascorbinsäure |
Stärkt das Immunsystem und die Blutgefäße. |
30 mg |
Vitamin B 1 |
Verbessert den Zustand des Nervensystems. |
0,3 mg |
Vitamin B 2 |
Sorgt für normale Stoffwechselprozesse. |
0,4 mg |
Vitamin B 5 |
Verantwortlich für den Hormonhaushalt und die Antikörperproduktion. |
2 mg |
Vitamin B 6 |
Verantwortlich für die Prozesse der Hämatopoese. |
0,5 mg |
Vitamin B 9 |
Verantwortlich für die Bildung neuer Zellstrukturen. |
25 µg |
Vitamin B 12 |
Verbessert die Funktion des Nervensystems. |
0,4 µg |
Vitamin PP |
Verantwortlich für Verdauungsprozesse. |
5 mg |
Vitamin H |
Verbessert die Leberfunktion. |
15 µg |
Tocopherol |
Stärkt die Blutgefäße. |
3 mg |
Vitamin K |
Normalisiert die Blutgerinnungsprozesse. |
10 µg |
Physiotherapeutische Behandlung
Physiotherapeutische Behandlungen führen im Allgemeinen bei keiner Form der Holoprosenzephalie zu signifikanten Ergebnissen.
Hausmittel
Es ist schwierig, über eine Volksbehandlung für eine so schwerwiegende Entwicklungsstörung eines Kindes wie die Holoprosenzephalie zu sprechen. Diese Krankheit ist so schwerwiegend, dass kranke Kinder in den meisten Fällen nicht einmal sechs Monate leben – und nur in seltenen Fällen kann ihre Existenz mit Hilfe einer chirurgischen Behandlung verlängert werden.
Eine Kräuterbehandlung in Form individueller Volksrezepte kann nur symptomatisch helfen: die Schwere der Krämpfe verringern, die Atmung erleichtern und die Funktion des Nervensystems des Babys normalisieren.
- Mischen Sie 1 Teil zerkleinerte Wermutsamen mit 4 Teilen Pflanzenöl und lassen Sie es über Nacht einwirken. Geben Sie dem Kind 1-2 Tropfen mit Zucker vermischt.
- 15 g Thymian in einem Glas kochendem Wasser aufbrühen und dem Patienten dreimal täglich 1 EL davon anbieten.
- Die Blütenblätter des Feldmohns werden getrocknet, zu Pulver gemahlen und in Milch gekocht. Honig wird hinzugefügt und dem Patienten wird im Laufe des Tages etwas davon verabreicht.
- Bereiten Sie eine Mischung aus 1 Teelöffel Anissamen, 1 Teelöffel Fenchelsamen, 1 Teelöffel Kümmelsamen und 2 Teelöffeln Minzblättern zu. Einen Esslöffel der Mischung in 200 ml kochendem Wasser aufbrühen, 30 Minuten ziehen lassen und abseihen. Geben Sie dem Patienten über den Tag verteilt kleine Mengen zu trinken.
- 2 Teelöffel Birkenknospen wie Tee aufbrühen. Zweimal täglich 100 ml trinken.
- Reiben Sie die Hände und Füße des Patienten mit Senföl ein.
- Geben Sie Honig zu Speisen und Getränken hinzu.
Pflanzen wie Maiglöckchen, Weiße Mistel, Baldrianwurzelstock, Walnussscheiden, Weißdorn- und Berberitzenfrüchte, Hopfenzapfen sowie Oregano, Thymian, Heidekraut und Steinklee haben eine gute krampflösende und stärkende Wirkung.
[ 39 ], [ 40 ], [ 41 ], [ 42 ], [ 43 ], [ 44 ]
Homöopathie
Homöopathische Arzneimittel gegen Holoprosenzephalie können nur von einem Arzt verschrieben werden. Wenn gleichzeitig traditionelle Arzneimittel verschrieben wurden, können diese nicht abgesetzt werden.
In der Regel wird eine 30-Cent-Verdünnung verwendet: Ein Körnchen wird in 100 ml Wasser verdünnt und dem Patienten wird täglich 1 Teelöffel davon eine halbe Stunde vor den Mahlzeiten verabreicht.
- Zincum metallicum – gegen Krampfanfälle im Kindesalter.
- Veratrum album – bei steifen Gelenken, Muskelsteifheit.
- Stramonium – gegen Demenz und Krampfanfälle.
- Stannum metallicum – gegen Krämpfe und Krämpfe.
- Plumbum metallicum – bei Muskelkrämpfen, Neurosen.
- Moshus – bei Krämpfen, Bewusstlosigkeit.
Psychologische Hilfe bei Holoprosenzephalie
Die Schwangerschaft ist eine wichtige und verantwortungsvolle Phase im Leben einer Frau. Die Diagnose „fetale Holoprosenzephalie“, die fast zu Beginn der Schwangerschaft gestellt wird, bringt der werdenden Mutter jedoch viele Ängste und Sorgen mit sich und stellt sie vor eine schwierige Entscheidung: sich für eine Abtreibung zu entscheiden oder ein Kind zur Welt zu bringen (was ist, wenn sich die Diagnose als falsch herausstellt)? In solchen Momenten macht sich eine Frau nicht nur Sorgen um die Gesundheit des sich im Mutterleib entwickelnden Babys, sondern auch darum, ob das Problem bei nachfolgenden Schwangerschaften erneut auftritt, ob sie überhaupt Kinder bekommen kann usw.
Manchmal macht die entstandene Situation der Schwangeren so große Angst, dass sich ihre Angehörigen ernsthaft Sorgen um ihren psychischen Zustand machen. In solchen Momenten ist es besser, einen Psychologen oder Psychotherapeuten aufzusuchen. Ein erfahrener Psychologe kann einer Frau in einer so schwierigen Zeit Unterstützung bieten, Harmonie und Vertrauen in die Zukunft wiederherstellen. Bei Bedarf kann allen Familienmitgliedern psychologische Hilfe angeboten werden.
Verhütung
Um die Entwicklung einer Holoprosenzephalie bei einem Baby zu verhindern, werden bereits während der Schwangerschaftsplanung und -empfängnis vorbeugende Maßnahmen ergriffen. Eine Frau, die sich auf die Rolle einer Mutter vorbereitet, sollte sehr auf ihre Gesundheit achten. Bereits vor der Schwangerschaft sollten alle bestehenden Erkrankungen behandelt und ein Zahnarzt, ein Gynäkologe und ein Genetiker konsultiert werden.
Sowohl während der Babyplanung als auch bereits während der Schwangerschaft ist es strengstens verboten, Alkohol zu trinken, zu rauchen, Drogen zu nehmen oder nicht ärztlich verschriebene Medikamente einzunehmen. Der behandelnde Arzt wiederum muss wissen, dass die Frau eine Schwangerschaft plant: Auf dieser Grundlage verschreibt er Medikamente, die die Qualität der Empfängnis und die Entstehung von Defekten beim Fötus nicht beeinträchtigen.
Eine schwangere Frau muss ab den ersten Tagen der Schwangerschaft ihre Gesundheit schützen und darf keinen Stress und keine übermäßige Belastung zulassen. Wichtig ist auch, sich rechtzeitig bei einer Frauensprechstunde anzumelden, die notwendigen Untersuchungen durchzuführen und auf die Empfehlungen eines Facharztes zu hören.
Prognose
Holoprosenzephalie hat eine äußerst ungünstige Prognose, deren Ausmaß vom Schweregrad der festgestellten Defekte abhängt. Die Sterblichkeitsrate hängt von der Art der Erkrankung ab. Bei vielen Frauen mit fetaler Holoprosenzephalie endet die Schwangerschaft mit einem spontanen Abbruch – der menschliche Körper entscheidet also selbst, dass das zukünftige Baby nicht existieren kann. Die meisten Babys mit Holoprosenzephalie sterben im ersten Lebensjahr. Bei mittelschweren Entwicklungsstörungen besteht die Chance, dass die Patientin das Säuglingsalter überlebt und die Pubertät erreicht.