
Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
Lissencephalie des Gehirns
Facharzt des Artikels

Zuletzt überprüft: 12.07.2025
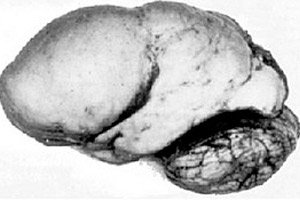
Unter den organischen Hirnerkrankungen sticht eine angeborene Anomalie der Gehirnentwicklung wie die Lissenzephalie hervor, deren Wesen in der fast glatten Oberfläche der Hirnrinde ihrer Hemisphären liegt – mit einer unzureichenden Anzahl von Windungen und Furchen. [ 1 ]
Bei völligem Fehlen von Windungen spricht man von Agyrie, das Vorhandensein mehrerer breiter, flacher Windungen von Pachygyrie. Diese Defekte haben wie einige andere Reduktionsdeformationen des Gehirns den Code Q04.3 in ICD-10.
Epidemiologie
Laut Statistik über seltene Erkrankungen gibt es 1-1,2 Fälle von Lissenzephalie pro 100.000 Neugeborenen. [ 2 ], [ 3 ]
Einigen Daten zufolge werden bei Kindern mit Miller-Dieker-Syndrom bis zu 25–30 % der Fälle einer klassischen Lissenzephalie beobachtet; bei fast 85 % der Patienten werden Punktmutationen und Deletionen der Gene LIS1 und DCX festgestellt. [ 4 ]
Genetische Studien von 17 Genen, die mit Lissenzephalie in Zusammenhang stehen, zeigten, dass 40 % der Patienten an einer Mutation oder Deletion von LIS1 leiden und 23 % mit einer DCX-Mutation in Zusammenhang stehen, gefolgt von TUBA1A (5 %) und DYNC1H1 (3 %).[ 5 ]
Ursachen Lissenzephalie
Alle bekannten Ursachen für die Bildung der Großhirnrinde (Cortex cerebri) fast oder vollständig ohne Windungen und Furchen, die den „Arbeitsbereich“ des menschlichen Gehirns vergrößern und die „Produktivität“ des Zentralnervensystems sicherstellen, sind mit Störungen in seiner perinatalen Entwicklung verbunden. Das heißt, beim Fötus entwickelt sich eine Lissenzephalie. [ 6 ]
Das Versagen bei der Bildung von Schichten der Großhirnrinde des Fötus bei Lissenzephalie ist das Ergebnis einer abnormalen Migration der Neuronen, die sie bilden, oder eines vorzeitigen Abbruchs dieses Prozesses.
Dieser für die zerebrokortikale Histogenese essentielle Prozess verläuft in mehreren Stadien von der 7. bis zur 18. Schwangerschaftswoche. Aufgrund der erhöhten Empfindlichkeit gegenüber genetischen Mutationen sowie verschiedenen negativen physikalischen, chemischen und biologischen Einflüssen kann jede Abweichung von der Norm zu einer falschen Lokalisierung von Neuronen mit der möglichen Bildung einer verdickten Schicht grauer Substanz des Kortex ohne charakteristische Struktur führen. [ 7 ]
In einigen Fällen ist Lissenzephalie bei Kindern mit dem Miller-Dieker-, Walker-Warburg- oder Norman-Roberts-Syndrom verbunden.
Lesen Sie auch – Entwicklungsstörungen des Gehirns
Risikofaktoren
Zu den Risikofaktoren für die Geburt eines Kindes mit einem so schweren Defekt zählen neben Mutationen in einigen Genen Sauerstoffmangel (Hypoxie) des Fötus, unzureichende Blutversorgung des Gehirns (Hypoperfusion), akuter zerebrovaskulärer Unfall in Form eines perinatalen Schlaganfalls, Plazentaerkrankungen, Virusinfektionen der schwangeren Frau (einschließlich TORCH), [ 8 ] Probleme mit dem allgemeinen Stoffwechsel und der Schilddrüsenfunktion, Rauchen, Alkohol, Psychopharmaka und Betäubungsmittel, Einnahme einer Reihe von Medikamenten und erhöhte Strahlenwerte. [ 9 ]
Pathogenese
Nicht alle Fälle von Lissenzephalie haben eine Pathogenese, die durch Chromosomenanomalien und Genmutationen verursacht wird. Es sind jedoch einige Gene bekannt, die Proteine kodieren, die eine wichtige Rolle bei der korrekten Bewegung von Neuroblasten und Neuronen entlang der radialen Gliazellen spielen – zur Bildung der Großhirnrinde. Und Mutationen dieser Gene führen zu dieser Pathologie. [ 10 ]
Insbesondere handelt es sich dabei um sporadische Mutationen (ohne Vererbung) des LIS1-Gens auf Chromosom 17, das das zytoplasmatische Motorprotein der Mikrotubuli Dynein reguliert, sowie des DCX-Gens auf dem X-Chromosom, das für das Protein Doublecortin (Lissencephalin-X) kodiert. [ 11 ]. Im ersten Fall definieren Spezialisten die klassische Lissenzephalie (Typ I), im zweiten – X-chromosomal. [ 12 ]
Wenn das FLN1-Gen, das das Phosphoprotein Filamin 1 kodiert, gelöscht wird, kann es sein, dass der Prozess der gerichteten neuronalen Migration überhaupt nicht beginnt, was zu einem vollständigen Fehlen von Windungen (Agyrie) führt. [ 13 ]
Es wurden Mutationen im CDK5-Gen identifiziert, das für ein Kinase-Enzym kodiert – einen Katalysator für den intrazellulären Stoffwechsel, der den Zellzyklus in ZNS-Neuronen reguliert und ihre normale Migration während der pränatalen Bildung von Gehirnstrukturen sicherstellt.
Abnorme Veränderungen im RELN-Gen auf Chromosom 7, die kortikale Gyrusdefekte beim Norman-Roberts-Syndrom verursachen, führen zu einem Mangel des extrazellulären Glykoproteins Reelin, das für die Regulierung der Migration und Positionierung neuraler Stammzellen während der Entwicklung des Cortex cerebri erforderlich ist.[ 14 ], [ 15 ], [ 16 ]
Das ARX-Gen kodiert das Non-Aristalens-Homöobox-Protein, einen Transkriptionsfaktor, der im Vorderhirn und anderen Geweben eine wichtige Rolle spielt.[ 17 ] Kinder mit der ARX-Mutation haben weitere Symptome, wie fehlende Teile des Gehirns (Agenesie des Corpus callosum), abnormale Genitalien und schwere Epilepsie.[ 18 ],[ 19 ]
Mehrere Gene wurden mit Lissenzephalie in Verbindung gebracht. Zu diesen Genen gehören VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A und CDK5.[ 20 ]
Das Cytomegalievirus (CMV) wird mit der Entwicklung einer Lissenzephalie in Verbindung gebracht, die auf eine verminderte Blutversorgung des fetalen Gehirns zurückzuführen ist. Der Schweregrad der CMV-Infektion hängt vom Gestationsalter ab. Eine frühe Infektion führt häufiger zu einer Lissenzephalie, da die neuronale Migration früh in der Schwangerschaft erfolgt.[ 21 ]
Darüber hinaus umfasst der Mechanismus des Auftretens dieser Anomalie eine unvollständige oder spätere Beendigung der Migration von Neuronen aus der periventrikulären generativen Zone zur Großhirnrinde. In solchen Fällen entwickelt sich entweder eine unvollständige Lissenzephalie oder eine Pachygyrie, bei der mehrere breite Rillen und Windungen gebildet werden (die meisten davon fehlen jedoch).
Symptome Lissenzephalie
Die ersten Anzeichen dieser Pathologie (ohne die zuvor genannten Syndrome) treten möglicherweise nicht unmittelbar nach der Geburt auf, sondern nach eineinhalb bis zwei Monaten. Am häufigsten werden die folgenden klinischen Symptome einer Lissenzephalie beobachtet:
- Muskelhypotonie, oft kombiniert mit spastischer Lähmung;
- Krämpfe und generalisierte tonisch-klonische Anfälle (in Form von Opisthotonus);
- schwere geistige Behinderung und Wachstumsverzögerung;
- Beeinträchtigung neurologischer und motorischer Funktionen.
Schluckbeschwerden können zu Schwierigkeiten beim Füttern des Babys führen. [ 22 ]
Eine hochgradige neuromotorische Beeinträchtigung äußert sich häufig in einer Tetraplegie – einer Lähmung aller Gliedmaßen. Deformationen der Hände, Finger oder Zehen sind möglich.
Beim Norman-Roberts-Syndrom mit Lissenzephalie Typ I werden kraniofaziale Anomalien beobachtet: schwere Mikrozephalie, niedrige Stirnneigung und hervorstehender breiter Nasenrücken, weit auseinanderstehende Augen (Hyperterlorismus), Unterentwicklung der Kiefer (Mikrognathie). [ 23 ]
Das Miller-Dieker-Syndrom kann außerdem durch einen ungewöhnlich kleinen Kopf mit breiter, hoher Stirn und kurzer Nase, Einsenkungen in den Schläfen (bitemporale Einsenkungen) und tief angesetzte, deformierte Ohren gekennzeichnet sein.
Das schwere Lissenzephalie-Syndrom ist durch Mikrozephalie gekennzeichnet, eine Verkleinerung der Augäpfel (Mikrophthalmie), kombiniert mit Netzhautdysplasie, obstruktivem Hydrozephalus und fehlendem oder hypoplastischem Corpus callosum.
Komplikationen und Konsequenzen
Zu den Komplikationen dieser Anomalie zählen laut Experten eine Beeinträchtigung der Schluckfunktion (Dysphagie) und gastroösophagealer Reflux, refraktäre (unkontrollierte) Epilepsie, häufige Infektionen der oberen Atemwege und Lungenentzündung (einschließlich chronischer Aspiration).
Säuglinge mit Lissenzephalie können angeborene organische Herzprobleme in Form eines Vorhofseptumdefekts oder eines komplexen Herzfehlers mit Zyanose (Fallot-Tetralogie) haben. [ 24 ]
Die Folgen einer postnatalen Wachstumsverzögerung führen in den meisten Fällen innerhalb von 24 Monaten nach der Geburt zum Tod.
Diagnose Lissenzephalie
Die Diagnose beginnt mit einer körperlichen Untersuchung des Kindes, einer Untersuchung der Krankengeschichte der Eltern sowie der Schwangerschafts- und Geburtsgeschichte.
Während der Schwangerschaft können zellfreie fetale DNA-Tests, Amniozentesen oder Chorionzottenbiopsien erforderlich sein. [ 25 ] Weitere Informationen finden Sie unter – Pränatale Diagnostik angeborener Erkrankungen
Die instrumentelle Diagnostik dient der Visualisierung von Gehirnstrukturen und der Beurteilung ihrer Funktionen:
- Computertomographie des Gehirns;
- Magnetresonanztomographie (MRT) des Gehirns;
- Elektroenzephalogramm (EEG). [ 26 ]
Während der Schwangerschaft kann bei der Ultraschalluntersuchung des Fötus nach der 20.–21. Woche aufgrund des Fehlens der parieto-okzipitalen und calcarinen Furchen sowie einer Anomalie der Sylvischen Fissur des Gehirns eine Lissenzephalie vermutet werden.
Differenzialdiagnose
Es wird eine Differentialdiagnose zu anderen Syndromen angeborener Hirndefekte durchgeführt.
Es gibt mehr als 20 Arten von Lissenzephalie, von denen die meisten in zwei Hauptkategorien eingeteilt werden: klassische Lissenzephalie (Typ 1) und Kopfsteinpflaster-Lissenzephalie (Typ 2). Jede Kategorie weist ähnliche klinische Manifestationen, aber unterschiedliche genetische Mutationen auf.[ 27 ]
Die Gehirnuntersuchung bei Lissenzephalie Typ I zeigt eine Großhirnrinde mit vier Schichten statt der sechs bei normalen Patienten. Bei Lissenzephalie Typ 2 hingegen ist die Großhirnrinde desorganisiert und erscheint knotig oder knotig. Grund dafür ist die vollständige Verdrängung der Großhirnrinde durch Ansammlungen kortikaler Neuronen, die durch gliomesenchymales Gewebe getrennt sind. Die Patienten wiesen zudem Muskel- und Augenanomalien auf.
- Klassische Lissenzephalie (Typ 1):
- LIS1: Isolierte Lissenzephalie und Miller-Dieker-Syndrom (Lissenzephalie in Verbindung mit Gesichtsdysmorphien). [ 28 ]
- LISX1: Mutation des DCX-Gens. Im Vergleich zur durch LIS1-Mutationen verursachten Lissenzephalie weist DCX einen sechsschichtigen Kortex statt vier auf.
- Isolierte Lissenzephalie ohne andere bekannte genetische Defekte
- Kopfsteinpflaster-Lissenzephalie (Typ 2):
- Walker-Warburg-Syndrom
- Fukuyama-Syndrom
- Erkrankungen der Muskeln, Augen und des Gehirns
- Andere Typen können nicht in eine der beiden oben genannten Gruppen eingeordnet werden:
- LIS2: Norman-Roberts-Syndrom, ähnlich der Lissenzephalie Typ I oder dem Miller-Dieker-Syndrom, jedoch ohne Deletion des Chromosoms 17.
- LIS3
- LISX2
Mikrolissenzephalie: Hierbei handelt es sich um eine Kombination aus dem Fehlen der normalen Rindenfaltung und einem ungewöhnlich kleinen Kopf. Kinder mit normaler Lissenzephalie haben bei der Geburt einen normal großen Kopf. Bei Kindern mit einem kleinen Kopf bei der Geburt wird in der Regel Mikrolissenzephalie diagnostiziert.
Es ist auch wichtig, zwischen Lissenzephalie und Polymikrogyrie zu unterscheiden, bei denen es sich um unterschiedliche Entwicklungsstörungen des Gehirns handelt.
Wen kann ich kontaktieren?
Behandlung Lissenzephalie
Da es sich bei der Lissenzephalie um einen unheilbaren organischen Defekt handelt, ist nur eine unterstützende und symptomatische Behandlung möglich. [ 29 ]
Dies ist in erster Linie die Verwendung von Antikonvulsiva und Antiepileptika sowie die Installation einer Gastrostomiesonde im Magen (wenn das Kind nicht selbstständig schlucken kann). Massage ist nützlich.
Bei einem schweren Hydrozephalus wird die Gehirn-Rückenmarks-Flüssigkeit entnommen.
Verhütung
Experten empfehlen werdenden Eltern eine genetische Beratung in Anspruch zu nehmen, Schwangeren rechtzeitig eine Anmeldung bei Frauenärzten und Geburtshelfern sowie alle vorgesehenen Untersuchungen durchzuführen.
Prognose
Bei Kindern mit Lissenzephalie hängt die Prognose vom Grad der Erkrankung ab. Meistens überschreitet die geistige Entwicklung des Kindes jedoch nicht das Niveau von vier bis fünf Monaten. Alle Kinder mit dieser Diagnose leiden an schweren psychomotorischen Störungen und schwer behandelbarer Epilepsie. [ 30 ]
Laut NINDS (dem amerikanischen National Institute of Neurological Disorders and Stroke) beträgt die maximale Lebenserwartung von Menschen mit Lissenzephalie etwa 10 Jahre.