
Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
Hereditäre Nephritis (Alport-Syndrom) bei Kindern
Facharzt des Artikels

Zuletzt überprüft: 05.07.2025
Die hereditäre Nephritis (Alport-Syndrom) ist eine genetisch bedingte hereditäre nicht-immunologische Glomerulopathie, die sich durch Hämaturie (manchmal mit Proteinurie), fortschreitende Abnahme der Nierenfunktion mit der Entwicklung eines chronischen Nierenversagens, oft verbunden mit sensorineuraler Taubheit und Sehbehinderung, manifestiert.
Die Krankheit wurde erstmals 1902 von L. G. Guthrie beschrieben, der eine Familie beobachtete, in der Hämaturie über mehrere Generationen hinweg auftrat. 1915 beschrieb A. F. Hurst die Entwicklung einer Urämie bei Mitgliedern derselben Familie. 1927 stellte A. Alport erstmals einen Hörverlust bei mehreren Verwandten mit Hämaturie fest. In den 1950er Jahren wurden Augenläsionen bei einer ähnlichen Erkrankung beschrieben. 1972 zeigten Hinglais et al. bei Patienten mit hereditärer Hämaturie bei einer morphologischen Untersuchung des Nierengewebes eine ungleichmäßige Ausdehnung und Schichtung der glomerulären Basalmembranen. 1985 wurde die genetische Grundlage der hereditären Nephritis identifiziert – eine Mutation im Typ-IV-Kollagen-Gen (Fiengold et al., 1985).
Die Untersuchung der genetischen Natur der Erkrankung ließ den Schluss zu, dass die Unterschiede in den phänotypischen Manifestationen der hereditären Nephritis (mit oder ohne Hörverlust) auf den Expressionsgrad des mutierten Gens zurückzuführen sind. Daher werden derzeit alle klinischen Varianten als Manifestationen einer Krankheit betrachtet, und der Begriff „hereditäre Nephritis“ ist synonym mit dem Begriff „Alport-Syndrom“.
Epidemiologischen Studien zufolge tritt die hereditäre Nephritis mit einer Häufigkeit von 17 pro 100.000 Kindern auf.
 [
[ Ursachen des Alport-Syndroms
Die genetische Grundlage der Erkrankung ist eine Mutation im Gen der a-5-Kette des Typ-IV-Kollagens. Dieser Typ ist universell für die Basalmembranen der Niere, des Cochlea-Apparats, der Linsenkapsel, der Netzhaut und der Hornhaut des Auges, was in Studien mit monoklonalen Antikörpern gegen diese Kollagenfraktion nachgewiesen wurde. Kürzlich wurde die Möglichkeit des Einsatzes von DNA-Sonden zur pränatalen Diagnostik der hereditären Nephritis aufgezeigt.
Es wird betont, wie wichtig es ist, alle Familienmitglieder mit DNA-Sonden zu testen, um Träger des mutierten Gens zu identifizieren. Dies ist für die medizinische und genetische Beratung von Familien mit dieser Krankheit von großer Bedeutung. Bis zu 20 % der Familien haben jedoch keine nierenkranken Verwandten, was auf eine hohe Häufigkeit spontaner Mutationen des abnormalen Gens hindeutet. Die meisten Patienten mit hereditärer Nephritis haben Personen mit Nierenerkrankungen, Hörverlust und Sehstörungen in ihren Familien. Blutsverwandte Ehen zwischen Personen mit einem oder mehreren Vorfahren sind wichtig, da bei der Ehe verwandter Personen die Wahrscheinlichkeit steigt, die gleichen Gene von beiden Elternteilen zu erhalten. Es wurden autosomal-dominante, autosomal-rezessive und dominante, X-chromosomale Übertragungswege etabliert.
Bei Kindern unterscheidet man am häufigsten drei Arten der hereditären Nephritis: das Alport-Syndrom, die hereditäre Nephritis ohne Hörverlust und die familiäre benigne Hämaturie.
Das Alport-Syndrom ist eine erbliche Nephritis mit Hörbehinderung. Es beruht auf einem kombinierten Defekt in der Struktur des Kollagens der glomerulären Basalmembran der Nieren-, Ohr- und Augenstrukturen. Das Gen des klassischen Alport-Syndroms befindet sich im Locus 21-22 q des langen Arms des X-Chromosoms. In den meisten Fällen wird es dominant vererbt und ist an das X-Chromosom gebunden. In dieser Hinsicht ist das Alport-Syndrom bei Männern schwerer ausgeprägt, da bei Frauen die Funktion des mutierten Gens durch ein gesundes Allel des zweiten, unbeschädigten Chromosoms kompensiert wird.
Die genetische Grundlage für die Entwicklung einer hereditären Nephritis sind Mutationen in den Genen der Alpha-Ketten des Kollagens Typ IV. Es sind sechs Alpha-Ketten des Kollagens Typ IV G bekannt: Die Gene der a5- und a6-Ketten (Col4A5 und Col4A5) befinden sich auf dem langen Arm des X-Chromosoms in der Zone 21-22q; die Gene der a3- und a4-Ketten (Col4A3 und Col4A4) befinden sich auf dem 2. Chromosom; die Gene der a1- und a2-Ketten (Col4A1 und Col4A2) befinden sich auf dem 13. Chromosom.
In den meisten Fällen (80–85 %) liegt ein X-chromosomaler Erbgang vor, der mit einer Schädigung des Col4A5-Gens infolge von Deletion, Punktmutationen oder Spleißstörungen einhergeht. Aktuell sind über 200 Mutationen des Col4A5-Gens bekannt, die für die Störung der Synthese der a5-Ketten von Kollagen Typ IV verantwortlich sind. Bei dieser Vererbung tritt die Erkrankung bei Kindern beiderlei Geschlechts auf, bei Jungen verläuft sie jedoch schwerer.
Mutationen in den Genloci der Gene Col4A3 und Col4A4, die für die Synthese der a3- und a4-Ketten von Typ-IV-Kollagen verantwortlich sind, werden autosomal vererbt. Studien zufolge wird der autosomal-dominante Vererbungstyp bei 16 % der Fälle von hereditärer Nephritis und der autosomal-rezessive Vererbungstyp bei 6 % der Patienten beobachtet. Es sind etwa 10 Mutationsvarianten der Gene Col4A3 und Col4A4 bekannt.
Das Ergebnis von Mutationen ist eine Verletzung der Montageprozesse von Kollagen Typ IV, was zu einer Verletzung seiner Struktur führt. Kollagen Typ IV ist einer der Hauptbestandteile der glomerulären Basalmembran, des Cochlea-Apparats und der Augenlinse, deren Pathologie in der Klinik für hereditäre Nephritis festgestellt wird.
Kollagen Typ IV, das Teil der glomerulären Basalmembran ist, besteht hauptsächlich aus zwei a1-Ketten (IV) und einer a2-Kette (IV) und enthält außerdem a3-, a4- und a5-Ketten. Bei der X-chromosomalen Vererbung geht die Mutation des Col4A5-Gens am häufigsten mit dem Fehlen von a3-, a4-, a5- und a6-Ketten in der Struktur von Kollagen Typ IV einher, während die Anzahl der o1- und a2-Ketten in der glomerulären Basalmembran zunimmt. Der Mechanismus dieses Phänomens ist unklar; es wird angenommen, dass die Ursache posttranskriptionelle Veränderungen der mRNA sind.
Das Fehlen von a3-, a4- und a5-Ketten in der Struktur des Kollagens Typ IV der glomerulären Basalmembranen führt zu deren Ausdünnung und Brüchigkeit in den frühen Stadien des Alport-Syndroms, das sich klinisch häufiger durch Hämaturie (seltener durch Hämaturie mit Proteinurie oder nur Proteinurie), Hörverlust und Lenticonus manifestiert. Ein weiteres Fortschreiten der Erkrankung führt in den späten Stadien der Erkrankung zu einer Verdickung und beeinträchtigten Durchlässigkeit der Basalmembranen mit einer Proliferation der Kollagentypen V und VI, was sich in einer Zunahme der Proteinurie und einer Abnahme der Nierenfunktion äußert.
Die Art der Mutation, die der hereditären Nephritis zugrunde liegt, bestimmt weitgehend ihre phänotypische Manifestation. Bei Deletion des X-Chromosoms mit gleichzeitiger Mutation der Gene Col4A5 und Col4A6, die für die Synthese der a5- und a6-Ketten von Kollagen Typ IV verantwortlich sind, ist das Alport-Syndrom mit einer Leiomyomatose der Speiseröhre und der Genitalien kombiniert. Forschungsdaten zufolge wird bei einer mit einer Deletion verbundenen Mutation des Col4A5-Gens im Vergleich zu einer Punktmutation dieses Gens ein schwererer pathologischer Prozess, eine Kombination von Nierenschäden mit extrarenalen Manifestationen und die frühe Entwicklung eines chronischen Nierenversagens festgestellt.
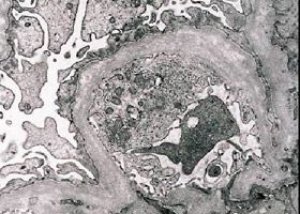
Morphologisch zeigt die Elektronenmikroskopie eine Ausdünnung und Schichtung der glomerulären Basalmembranen (insbesondere der Lamina densa) sowie das Vorhandensein elektronendichter Granula. Glomeruläre Läsionen können bei demselben Patienten heterogen sein, von minimalen fokalen mesangialen Läsionen bis hin zu Glomerulosklerose. Die Glomerulitis beim Alport-Syndrom ist stets immunnegativ, was sie von der Glomerulonephritis unterscheidet. Charakteristische Merkmale sind die Entwicklung einer tubulären Atrophie, lymphohistiozytäre Infiltration und das Vorhandensein von „Schaumzellen“ mit Lipideinschlüssen – Lipophagen. Im weiteren Krankheitsverlauf zeigen sich eine Verdickung und ausgeprägte Zerstörung der glomerulären Basalmembranen.
Es zeigen sich Veränderungen im Immunsystem. Patienten mit hereditärer Nephritis haben einen erniedrigten IgA-Spiegel und eine Tendenz zu einem Anstieg der IgM-Konzentration im Blut. Der IgG-Spiegel kann in den frühen Stadien der Erkrankung erhöht sein und in den späteren Stadien sinken. Möglicherweise ist der Anstieg der IgM- und G-Konzentration eine Art kompensatorische Reaktion auf den IgA-Mangel.
Die funktionelle Aktivität des T-Lymphozytensystems ist reduziert; es kommt zu einer selektiven Abnahme der B-Lymphozyten, die für die Synthese von Ig A verantwortlich sind, die phagozytische Verbindung der Immunität ist gestört, hauptsächlich aufgrund einer Störung der Chemotaxis und der intrazellulären Verdauungsprozesse in Neutrophilen
Bei der Untersuchung einer Nierenbiopsie bei Patienten mit Alport-Syndrom zeigen elektronenmikroskopische Daten ultrastrukturelle Veränderungen der glomerulären Basalmembran: Ausdünnung, Strukturstörung und Spaltung der glomerulären Basalmembranen mit Veränderung ihrer Dicke und ungleichmäßigen Konturen. In den frühen Stadien der hereditären Nephritis bestimmt der Defekt die Ausdünnung und Brüchigkeit der glomerulären Basalmembranen.
Eine Ausdünnung der glomerulären Membranen ist ein günstigeres Zeichen und tritt häufiger bei Mädchen auf. Ein konstanteres elektronenmikroskopisches Zeichen bei hereditärer Nephritis ist die Spaltung der Basalmembran, wobei der Schweregrad ihrer Zerstörung mit der Schwere des Prozesses korreliert.
Symptome des Alport-Syndroms bei Kindern
Die ersten Symptome des Alport-Syndroms in Form eines isolierten Harnwegssyndroms treten am häufigsten bei Kindern der ersten drei Lebensjahre auf. In den meisten Fällen wird die Krankheit zufällig entdeckt. Das Harnwegssyndrom wird bei einer Vorsorgeuntersuchung des Kindes, vor der Aufnahme in eine Kindertagesstätte oder bei akuter respiratorischer Virusinfektion (ARVI) festgestellt. Im Falle einer Pathologie im Urin während einer ARVI. Bei hereditärer Nephritis gibt es im Gegensatz zur erworbenen Glomerulonephritis keine Latenzzeit.
Im Anfangsstadium der Erkrankung leidet der Gesundheitszustand des Kindes wenig, ein charakteristisches Merkmal ist die Persistenz und Resistenz des Harnsyndroms. Eines der Hauptsymptome ist Hämaturie unterschiedlichen Schweregrades, die in 100 % der Fälle beobachtet wird. Eine Zunahme des Hämaturiegrades wird während oder nach Atemwegsinfektionen, körperlicher Aktivität oder nach vorbeugenden Impfungen festgestellt. Die Proteinurie überschreitet in den meisten Fällen 1 g / Tag nicht, zu Beginn der Erkrankung kann sie inkonsistent sein, im weiteren Verlauf nimmt die Proteinurie zu. Periodisch kann eine Leukozyturie mit einem Überwiegen von Lymphozyten im Harnsediment vorhanden sein, die mit der Entwicklung interstitieller Veränderungen verbunden ist.
Anschließend ist die teilweise Nierenfunktion beeinträchtigt, der Allgemeinzustand des Patienten verschlechtert sich: Es treten Intoxikation, Muskelschwäche, arterielle Hypotonie, häufig Hörstörungen (insbesondere bei Jungen) und manchmal Sehstörungen auf. Eine Intoxikation äußert sich in Blässe, Müdigkeit und Kopfschmerzen. Im Anfangsstadium der Erkrankung wird ein Hörverlust meist nur durch eine Audiographie festgestellt. Ein Hörverlust beim Alport-Syndrom kann in verschiedenen Phasen der Kindheit auftreten, am häufigsten wird er jedoch im Alter von 6-10 Jahren diagnostiziert. Der Hörverlust bei Kindern beginnt mit hohen Frequenzen, erreicht einen signifikanten Grad in der Luft- und Knochenleitung und geht von einem Schallleitungs- zu einem Schallwahrnehmungs-Hörverlust über. Hörverlust kann eines der ersten Symptome der Erkrankung sein und einem Harnwegssyndrom vorausgehen.
In 20 % der Fälle weisen Patienten mit Alport-Syndrom Veränderungen der Sehorgane auf. Die am häufigsten festgestellten Anomalien betreffen die Linse: Sphärophokie, vorderer, hinterer oder gemischter Lentikonus und verschiedene Katarakte. In Familien mit Alport-Syndrom kommt Myopie häufig vor. Einige Forscher stellen in diesen Familien ständig bilaterale perimakuläre Veränderungen in Form von hellen weißlichen oder gelblichen Granulationen im Corpus luteum fest. Sie erachten dieses Zeichen als ein konstantes Symptom mit hohem diagnostischen Wert beim Alport-Syndrom. KS Chugh et al. (1993) stellten in einer ophthalmologischen Studie bei Patienten mit Alport-Syndrom eine Abnahme der Sehschärfe in 66,7 % der Fälle fest, einen vorderen Lentikonus in 37,8 %, Netzhautflecken in 22,2 %, Katarakte in 20 % und Keratokonus in 6,7 %.
Bei einigen Kindern mit hereditärer Nephritis, insbesondere bei Nierenversagen, ist eine erhebliche Verzögerung der körperlichen Entwicklung zu beobachten. Mit fortschreitendem Nierenversagen entwickelt sich eine arterielle Hypertonie. Bei Kindern wird sie häufiger in der Adoleszenz und in älteren Altersgruppen festgestellt.
Patienten mit hereditärer Nephritis sind durch das Vorhandensein verschiedener (mehr als 5-7) Narben der Bindegewebsdysmorphogenese gekennzeichnet. Zu den Bindegewebsstigmata bei Patienten zählen Hypertelorismus der Augen, hoher Gaumen, Bissanomalien, abnorme Form der Ohrmuscheln, Krümmung des kleinen Fingers an den Händen und „Sandalenlücke“ an den Füßen. Hereditäre Nephritis ist durch die Einheitlichkeit der Dysmorphogenese-Stigmata innerhalb einer Familie sowie eine hohe Häufigkeit ihrer Verbreitung unter Verwandten von Probanden gekennzeichnet, entlang deren Linie die Krankheit übertragen wird.
In den frühen Stadien der Erkrankung zeigt sich eine isolierte Abnahme partieller Nierenfunktionen: Transport von Aminosäuren, Elektrolyten, Konzentrationsfunktion, Azidogenese. Spätere Veränderungen beeinflussen den Funktionszustand sowohl des proximalen als auch des distalen Teils des Nephrons und sind durch kombinierte partielle Störungen gekennzeichnet. Eine Abnahme der glomerulären Filtration tritt später, häufiger in der Adoleszenz, auf. Mit fortschreitender hereditärer Nephritis entwickelt sich eine Anämie.
So ist die hereditäre Nephritis durch einen stufenweisen Krankheitsverlauf gekennzeichnet: zunächst ein latentes Stadium oder versteckte klinische Symptome, die sich in minimalen Veränderungen des Harnsyndroms manifestieren, dann eine allmähliche Dekompensation des Prozesses mit einer Abnahme der Nierenfunktion mit manifesten klinischen Symptomen (Intoxikation, Asthenie, Entwicklungsverzögerung, Anämie). Klinische Symptome treten in der Regel unabhängig von der Schichtung der Entzündungsreaktion auf.
Eine hereditäre Nephritis kann sich in verschiedenen Altersstufen manifestieren, was von der Wirkung des Gens abhängt, das bis zu einem bestimmten Zeitpunkt in einem unterdrückten Zustand ist.
Einstufung
Es gibt drei Arten von hereditärer Nephritis
- Option I – klinisch manifestiert sich eine Nephritis mit Hämaturie, Hörverlust und Augenschäden. Der Verlauf der Nephritis ist progressiv und führt zur Entwicklung eines chronischen Nierenversagens. Der Vererbungstyp ist dominant und an das X-Chromosom gebunden. Morphologisch zeigt sich eine Verletzung der Struktur der Basalmembran, deren Ausdünnung und Spaltung.
- Variante II – klinisch manifestiert sich die Nephritis als Hämaturie ohne Hörverlust. Der Verlauf der Nephritis ist progressiv und führt zur Entwicklung eines chronischen Nierenversagens. Der Vererbungstyp ist dominant und an das X-Chromosom gebunden. Morphologisch zeigt sich eine Ausdünnung der glomerulären Kapillarbasalmembran (insbesondere der Laminadensa).
- Option III – gutartige familiäre Hämaturie. Der Verlauf ist günstig, chronisches Nierenversagen entwickelt sich nicht. Die Vererbung ist autosomal-dominant oder autosomal-rezessiv. Beim autosomal-rezessiven Erbgang ist bei Frauen ein schwererer Krankheitsverlauf zu beobachten.
Diagnose des Alport-Syndroms
Folgende Kriterien werden vorgeschlagen:
- das Vorhandensein von mindestens zwei Patienten mit Nephropathie in jeder Familie;
- Hämaturie als Leitsymptom einer Nephropathie beim Probanden;
- das Vorliegen eines Hörverlusts bei mindestens einem Familienmitglied;
- Entwicklung eines chronischen Nierenversagens bei einem oder mehreren Verwandten.
Bei der Diagnostik verschiedener erblicher und angeborener Erkrankungen wird großer Wert auf eine umfassende Untersuchungsmethodik und vor allem auf die Berücksichtigung der bei der Erstellung des kindlichen Stammbaums gewonnenen Daten gelegt. Die Diagnose des Alport-Syndroms gilt als gültig, wenn beim Patienten 3 von 4 typischen Symptomen festgestellt werden: Hämaturie und chronisches Nierenversagen in der Familie, neurosensorischer Hörverlust, Sehstörungen beim Patienten, Nachweis von Anzeichen einer Spaltung der glomerulären Basalmembran mit Veränderung ihrer Dicke und ungleichmäßigen Konturen bei der elektronenmikroskopischen Untersuchung der Biopsie.
Die Untersuchung des Patienten sollte klinische und genetische Forschungsmethoden umfassen; gezielte Studie der Krankheitsgeschichte; allgemeine Untersuchung des Patienten unter Berücksichtigung diagnostisch relevanter Kriterien. In der Kompensationsphase kann eine Pathologie nur durch Konzentration auf Syndrome wie das Vorhandensein einer erblichen Belastung, Hypotonie, multiple Stigmata der Dysembryogenese, Veränderungen des Harnsyndroms erkannt werden. In der Dekompensationsphase können extrarenale Symptome auftreten, wie schwere Intoxikation, Asthenie, verzögerte körperliche Entwicklung, Anämie, die sich mit einer allmählichen Abnahme der Nierenfunktion manifestieren und verstärken. Bei den meisten Patienten mit einer Abnahme der Nierenfunktion wird Folgendes beobachtet: verminderte Acido- und Aminogenese; 50 % der Patienten stellen eine signifikante Abnahme der sekretorischen Funktion der Nieren fest; eingeschränkter Schwankungsbereich der optischen Dichte des Urins; Störung des Filtrationsrhythmus und dann eine Abnahme der glomerulären Filtration. Das Stadium des chronischen Nierenversagens wird diagnostiziert, wenn bei Patienten über einen Zeitraum von 3–6 Monaten oder länger ein erhöhter Harnstoffspiegel im Blutserum (mehr als 0,35 g/l) und eine Abnahme der glomerulären Filtration auf 25 % des Normalwerts vorliegt.
Die Differentialdiagnostik der hereditären Nephritis sollte primär mit der hämatologischen Form der erworbenen Glomerulonephritis in Verbindung gebracht werden. Die erworbene Glomerulonephritis beginnt meist akut, innerhalb von 2–3 Wochen nach der Infektion, mit extrarenalen Symptomen, einschließlich Hypertonie ab den ersten Tagen (bei hereditärer Nephritis hingegen Hypotonie), verminderter glomerulärer Filtration zu Beginn der Erkrankung und fehlender Beeinträchtigung partieller Tubulusfunktionen, während diese bei hereditären Nephritis vorhanden sind. Die erworbene Glomerulonephritis geht mit ausgeprägterer Hämaturie und Proteinurie sowie erhöhter BSG einher. Typische Veränderungen der glomerulären Basalmembran, charakteristisch für die hereditäre Nephritis, sind diagnostisch wertvoll.
Die Differentialdiagnose der dysmetabolischen Nephropathie erfolgt bei chronischem Nierenversagen, in der Familie klinisch nachgewiesenen heterogenen Nierenerkrankungen, und es kann ein Spektrum der Nephropathie von Pyelonephritis bis Urolithiasis geben. Kinder haben oft Beschwerden über Schmerzen im Bauch und regelmäßig beim Wasserlassen, im Urinsediment - Oxalate.
Bei Verdacht auf eine hereditäre Nephritis sollte zur Abklärung der Diagnose eine Überweisung an eine nephrologische Fachabteilung erfolgen.
Was muss untersucht werden?
Welche Tests werden benötigt?
Wen kann ich kontaktieren?
Behandlung des Alport-Syndroms
Das Behandlungsschema beinhaltet Einschränkungen bei schwerer körperlicher Anstrengung und Aufenthalt an der frischen Luft. Die Ernährung ist vollwertig und enthält ausreichend Proteine, Fette und Kohlenhydrate unter Berücksichtigung der Nierenfunktion. Von großer Bedeutung ist die Erkennung und Behandlung chronischer Infektionsherde. Folgende Medikamente werden eingesetzt: ATP, Cocarboxylase, Pyridoxin (bis zu 50 mg/Tag), Carnitinchlorid. Die Behandlungen finden 2-3 Mal jährlich statt. Bei Hämaturie werden pflanzliche Arzneimittel verschrieben – Brennnessel, Aroniasaft und Schafgarbe.
In der ausländischen und inländischen Literatur gibt es Berichte über die Behandlung mit Prednisolon und den Einsatz von Zytostatika. Die Wirkung lässt sich jedoch nur schwer beurteilen.
Bei chronischem Nierenversagen kommen Hämodialyse und Nierentransplantation zum Einsatz.
Es gibt keine Methoden zur spezifischen (wirksamen pathogenetischen) Therapie der hereditären Nephritis. Alle Behandlungsmaßnahmen zielen darauf ab, den Rückgang der Nierenfunktion zu verhindern und zu verlangsamen.
Die Ernährung sollte ausgewogen und kalorienreich sein und den Funktionszustand der Nieren berücksichtigen. Liegen keine Funktionsstörungen vor, sollte die Ernährung des Kindes ausreichend Proteine, Fette und Kohlenhydrate enthalten. Bei Anzeichen einer Nierenfunktionsstörung sollte die Menge an Protein, Kohlenhydraten, Kalzium und Phosphor begrenzt werden, um die Entwicklung eines chronischen Nierenversagens zu verzögern.
Körperliche Aktivitäten sollten eingeschränkt werden; Kindern wird geraten, Sport zu vermeiden.
Der Kontakt mit infektiösen Patienten sollte vermieden und das Risiko akuter Atemwegserkrankungen verringert werden. Die Sanierung chronischer Infektionsherde ist notwendig. Bei Kindern mit hereditärer Nephritis werden keine vorbeugenden Impfungen durchgeführt, eine Impfung ist nur bei epidemiologischen Indikationen möglich.
Hormonelle und immunsuppressive Therapien bei hereditärer Nephritis sind unwirksam. Es gibt Hinweise auf einen positiven Effekt (Verringerung der Proteinurie und Verlangsamung des Krankheitsverlaufs) bei mehrjähriger Langzeitanwendung von Ciclosporin A und ACE-Hemmern.
Bei der Behandlung von Patienten werden Medikamente eingesetzt, die den Stoffwechsel verbessern:
- Pyridoxin – 2–3 mg/kg/Tag in 3 Dosen über 4 Wochen;
- Cocarboxylase – 50 mg intramuskulär jeden zweiten Tag, insgesamt 10–15 Injektionen;
- ATP – 1 ml intramuskulär jeden zweiten Tag, 10–15 Injektionen;
- Vitamin A – 1000 IE/Jahr/Tag in 1 Dosis für 2 Wochen;
- Vitamin E – 1 mg/kg/Tag in 1 Dosis für 2 Wochen.
Diese Therapieform trägt zur Verbesserung des Allgemeinzustands der Patienten bei, verringert Funktionsstörungen der Eileiter und wird dreimal jährlich in Kursen durchgeführt.
Levamisol kann als Immunmodulator verwendet werden – 2 mg/kg/Tag 2-3 Mal pro Woche mit Pausen zwischen den Dosen von 3-4 Tagen.
Forschungsdaten zufolge hat die hyperbare Sauerstoffversorgung einen positiven Einfluss auf den Schweregrad von Hämaturie und Nierenfunktionsstörungen.
Die wirksamste Behandlungsmethode für hereditäre Nephritis ist eine rechtzeitige Nierentransplantation. In diesem Fall kommt es im Transplantat nicht zu einem Rückfall der Erkrankung; in einem kleinen Prozentsatz der Fälle (ca. 5 %) kann sich in der transplantierten Niere eine Nephritis entwickeln, die mit Antigenen der glomerulären Basalmembran assoziiert ist.
Eine vielversprechende Richtung ist die pränatale Diagnostik und die gentechnische Therapie. Tierversuche zeigen eine hohe Effizienz bei der Übertragung normaler Gene, die für die Synthese von Kollagen-Alpha-Ketten Typ IV verantwortlich sind, in das Nierengewebe, woraufhin die Synthese normaler Kollagenstrukturen beobachtet wird.
Vorhersage
Die Prognose einer hereditären Nephritis ist immer ernst.
Prognostisch ungünstige Kriterien für den Verlauf einer hereditären Nephritis sind:
- männliches Geschlecht;
- frühe Entwicklung eines chronischen Nierenversagens bei Familienmitgliedern;
- Proteinurie (mehr als 1 g/Tag);
- Verdickung der glomerulären Basalmembranen laut Mikroskopie;
- Akustikusneuritis;
- Deletion im Col4A5-Gen.
Die Prognose bei benigner familiärer Hämaturie ist günstiger.