
Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
Elektromyographie mit der Nadel
Facharzt des Artikels

Zuletzt überprüft: 06.07.2025
Die Nadelelektromyographie umfasst die folgenden Hauptmethoden:
- Standard-Nadel-EMG;
- EMG einzelner Muskelfasern;
- Makro-EMG;
- Scan-EMG.
Standard-Nadel-Elektromyographie
Die Nadelelektromyographie ist eine invasive Untersuchungsmethode, die mit Hilfe einer konzentrischen Nadelelektrode durchgeführt wird, die in den Muskel eingeführt wird. Die Nadelelektromyographie ermöglicht die Beurteilung des peripheren neuromotorischen Apparats: der morphofunktionellen Organisation der motorischen Einheiten der Skelettmuskulatur, des Zustands der Muskelfasern (ihrer spontanen Aktivität) und im Falle einer dynamischen Beobachtung die Beurteilung der Wirksamkeit der Behandlung, der Dynamik des pathologischen Prozesses und der Prognose der Erkrankung.
Diagnosewert
Die Standardnadel-Elektromyographie nimmt einen zentralen Platz unter den elektrophysiologischen Untersuchungsmethoden bei verschiedenen neuromuskulären Erkrankungen ein und ist von entscheidender Bedeutung für die Differentialdiagnose neurogener und primärer Muskelerkrankungen.
Mit dieser Methode lässt sich der Schweregrad der Denervierung des vom betroffenen Nerv innervierten Muskels, der Grad seiner Erholung und die Wirksamkeit der Reinnervierung bestimmen.
Die Nadelelektromyographie findet nicht nur in der Neurologie Anwendung, sondern auch in der Rheumatologie, Endokrinologie, Sport- und Arbeitsmedizin, Pädiatrie, Urologie, Gynäkologie, Chirurgie und Neurochirurgie, Augenheilkunde, Zahnmedizin und Kieferchirurgie, Orthopädie und zahlreichen anderen medizinischen Bereichen.
Hinweise für das Verfahren
Erkrankungen der Motoneuronen des Rückenmarks ( ALS, spinale Amyotrophien, Poliomyelitis und Post-Polio-Syndrom, Syringomyelie usw.), Myelopathien, Radikulopathien, verschiedene Neuropathien (axonale und demyelinisierende), Myopathien, entzündliche Muskelerkrankungen ( Polymyositis und Dermatomyositis ), zentrale Bewegungsstörungen, Schließmuskelstörungen und eine Reihe anderer Situationen, in denen der Zustand der motorischen Funktionen und des Bewegungskontrollsystems objektiviert werden muss, um die Beteiligung verschiedener Strukturen des peripheren neuromotorischen Apparats an dem Prozess zu beurteilen.
 [ 6 ]
[ 6 ]
Vorbereitung
Der Patient benötigt keine besondere Vorbereitung auf die Untersuchung. Die Nadelelektromyographie erfordert eine vollständige Entspannung der zu untersuchenden Muskeln und wird daher im Liegen durchgeführt. Der Patient wird den zu untersuchenden Muskeln ausgesetzt, auf dem Rücken (oder Bauch) auf einer bequemen, weichen Liege mit verstellbarer Kopfstütze abgelegt, über die bevorstehende Untersuchung informiert und darüber, wie er den Muskel anspannen und wieder entspannen soll.
[ 7 ]
Technik Nadelelektromyographie
Die Untersuchung erfolgt mit einer konzentrischen Nadelelektrode, die in den motorischen Punkt des Muskels eingeführt wird (der zulässige Radius beträgt maximal 1 cm für große Muskeln und 0,5 cm für kleine). Die Potenziale der MU (PMU) werden aufgezeichnet. Bei der Auswahl der PMU für die Analyse sind bestimmte Regeln zu beachten.
Wiederverwendbare Nadelelektroden werden im Autoklaven oder durch andere Sterilisationsverfahren vorsterilisiert. Sterile Einweg-Nadelelektroden werden unmittelbar vor der Muskeluntersuchung geöffnet.
Nach dem Einführen der Elektrode in einen völlig entspannten Muskel und jeder Bewegung wird das mögliche Auftreten spontaner Aktivität überwacht. Die PMU wird bei minimaler willkürlicher Muskelanspannung aufgezeichnet, wodurch die Identifizierung einzelner PMUs ermöglicht wird. 20 verschiedene PMUs werden ausgewählt, wobei eine bestimmte Abfolge der Elektrodenbewegungen im Muskel beobachtet wird.
Bei der Beurteilung des Muskelzustands wird eine quantitative Analyse der festgestellten Spontanaktivität durchgeführt. Dies ist insbesondere für die Überwachung des Patientenzustands im Laufe der Zeit sowie für die Bestimmung der Therapiewirksamkeit wichtig. Die Parameter der aufgezeichneten Potenziale verschiedener motorischer Einheiten werden analysiert.
Nadelelektromyographie bei synaptischen Erkrankungen
Bei synaptischen Erkrankungen gilt die Nadelelektromyographie als zusätzliche Untersuchungsmethode. Bei Myasthenie ermöglicht sie die Beurteilung des Grades der „Blockierung“ der Muskelfasern in der MU, der durch das Ausmaß der Abnahme der durchschnittlichen Dauer der MU in den untersuchten Muskeln bestimmt wird. Das Hauptziel der Nadelelektromyographie bei Myasthenie ist jedoch der Ausschluss möglicher Begleiterkrankungen (Polymyositis, Myopathie, endokrine Störungen, verschiedene Polyneuropathien usw.). Die Nadelelektromyographie bei Patienten mit Myasthenie wird auch verwendet, um das Ausmaß der Reaktion auf die Verabreichung von Anticholinesterase-Medikamenten zu bestimmen, d. h. um die Veränderung der MU-Parameter bei Verabreichung von Neostigminmethylsulfat (Proserin) zu beurteilen. Nach der Einführung des Medikaments verlängert sich die Dauer der MU in den meisten Fällen. Das Ausbleiben einer Reaktion kann auf eine sogenannte myasthenische Myopathie hinweisen.
Die wichtigsten elektromyographischen Kriterien für synaptische Erkrankungen:
- Verkürzung der durchschnittlichen Dauer von PDE;
- Abnahme der Amplitude einzelner PMUs (kann fehlen);
- mäßige Polyphasie der PDE (kann fehlen);
- Fehlen spontaner Aktivität oder Vorhandensein von nur isoliertem PF.
Bei Myasthenie ist die durchschnittliche Dauer der MUAP in der Regel leicht reduziert (um 10–35 %). Die Mehrzahl der MUAPs weist eine normale Amplitude auf, jedoch werden in jedem Muskel mehrere MUAPs mit reduzierter Amplitude und Dauer registriert. Die Anzahl polyphasischer MUAPs überschreitet 15–20 % nicht. Spontane Aktivität fehlt. Bei ausgeprägter PF sollte eine Kombination von Myasthenie mit Hypothyreose, Polymyositis oder anderen Erkrankungen in Betracht gezogen werden.
Nadelelektromyographie bei primären Muskelerkrankungen
Die Nadelelektromyographie ist die wichtigste elektrophysiologische Methode zur Diagnose primärer Muskelerkrankungen (verschiedener Myopathien). Aufgrund der verminderten Fähigkeit der motorischen Einheiten, ausreichend Kraft für minimale Anstrengungen zu entwickeln, muss ein Patient mit einer primären Muskelerkrankung eine große Anzahl motorischer Einheiten rekrutieren. Dies bestimmt die Besonderheit der Elektromyographie bei solchen Patienten. Bei minimaler willkürlicher Muskelspannung ist es schwierig, einzelne motorische Einheiten zu isolieren; auf dem Bildschirm erscheint eine so große Anzahl kleiner Potentiale, dass ihre Identifizierung unmöglich ist. Dies ist das sogenannte myopathische Muster der Elektromyographie.
Bei entzündlichen Myopathien (Polymyositis) kommt es zu einem Reinnervationsprozess, der eine Erhöhung der Parameter des MUAP verursachen kann.
Die wichtigsten elektromyographischen Kriterien für primäre Muskelerkrankungen:
- Verkürzung der durchschnittlichen Dauer von PDE um mehr als 12 %;
- Abnahme der Amplitude einzelner PMUs (die durchschnittliche Amplitude kann entweder reduziert oder normal sein und manchmal auch erhöht);
- Polyphasie der PDE;
- ausgeprägte spontane Aktivität der Muskelfasern bei entzündlicher Myopathie (Polymyositis) oder PMD (in anderen Fällen ist sie minimal oder fehlt).
Eine Verkürzung der durchschnittlichen Dauer des MUAP ist ein Kardinalsymptom jeder primären Muskelerkrankung. Der Grund für diese Veränderung liegt darin, dass bei Myopathien Muskelfasern verkümmern und einige von ihnen aufgrund von Nekrose aus der MU-Zusammensetzung ausfallen, was zu einer Abnahme der MUAP-Parameter führt. Eine Verkürzung der Dauer der meisten MUAPs ist bei fast allen Muskeln von Patienten mit Myopathien nachweisbar, wobei sie in den klinisch am stärksten betroffenen proximalen Muskeln stärker ausgeprägt ist.
Das Histogramm der Verteilung der PMU nach Dauer verschiebt sich zu kleineren Werten (Stadium I oder II). Eine Ausnahme bildet die PMD: Aufgrund der ausgeprägten Polyphasie der PMU, die manchmal 100 % erreicht, kann die durchschnittliche Dauer deutlich erhöht sein.
Elektromyographie einzelner Muskelfasern
Die Elektromyographie einzelner Muskelfasern ermöglicht die Untersuchung der elektrischen Aktivität einzelner Muskelfasern, einschließlich der Bestimmung ihrer Dichte in Muskelmotoreinheiten und der Zuverlässigkeit der neuromuskulären Übertragung mithilfe der Jitter-Methode.
Für die Untersuchung wird eine spezielle Elektrode mit einer sehr kleinen Entladungsfläche von 25 µm Durchmesser benötigt, die sich 3 mm vom Ende entfernt an der Mantelfläche befindet. Die kleine Entladungsfläche ermöglicht die Aufzeichnung der Potenziale einer einzelnen Muskelfaser in einem Bereich mit einem Radius von 300 µm.
Studie zur Muskelfaserdichte
Die Bestimmung der Muskelfaserdichte in der MU basiert auf der Tatsache, dass der Bereich der Mikroelektrode zur Aufzeichnung der Aktivität einer einzelnen Muskelfaser streng definiert ist. Als Maß für die Muskelfaserdichte in der MU dient die durchschnittliche Anzahl der Potenziale einzelner Muskelfasern, die in ihrem Aufzeichnungsbereich während der Untersuchung von 20 verschiedenen MU in unterschiedlichen Muskelzonen aufgezeichnet wurden. Normalerweise kann diese Zone nur eine (selten zwei) Muskelfasern derselben MU enthalten. Durch eine spezielle methodische Technik (Triggergerät) kann die Anzeige von Potenzialen einzelner Muskelfasern anderer MU auf dem Bildschirm vermieden werden.
Die durchschnittliche Faserdichte wird in konventionellen Einheiten durch Berechnung der durchschnittlichen Anzahl von Potenzialen einzelner Muskelfasern verschiedener Muskelgruppen gemessen. Bei gesunden Menschen variiert dieser Wert je nach Muskel und Alter zwischen 1,2 und 1,8. Eine Zunahme der Muskelfaserdichte in Muskelgruppen spiegelt eine Veränderung der Muskelgruppenstruktur im Muskel wider.
Erforschung des Jitter-Phänomens
Normalerweise ist es immer möglich, die Elektrode zur Aufzeichnung einer einzelnen Muskelfaser in einem Muskel so zu positionieren, dass die Potenziale zweier benachbarter Muskelfasern einer motorischen Einheit aufgezeichnet werden. Wird das Potenzial der ersten Faser durch die Triggervorrichtung aktiviert, kommt es zu einer geringfügigen zeitlichen Verschiebung des Potenzials der zweiten Faser, da der Impuls unterschiedliche Zeit benötigt, um zwei unterschiedlich lange Nervenenden zu durchlaufen. Dies spiegelt sich in der Variabilität des Interpeak-Intervalls wider, d. h. die Aufzeichnungszeit des zweiten Potenzials schwankt im Verhältnis zum ersten, definiert als Potenzialtanz oder -jitter, dessen Wert normalerweise 5–50 μs beträgt.
Jitter spiegelt die Variabilität der neuromuskulären Übertragungszeit in zwei motorischen Endplatten wider. Daher ermöglicht diese Methode die Untersuchung der Stabilität der neuromuskulären Übertragung. Bei Störungen durch eine Pathologie nimmt der Jitter zu. Der ausgeprägteste Anstieg wird bei synaptischen Erkrankungen, vor allem bei Myasthenie, beobachtet.
Bei einer deutlichen Verschlechterung der neuromuskulären Übertragung liegt ein Zustand vor, bei dem ein Nervenimpuls eine von zwei benachbarten Fasern nicht erregen kann und es zu einer sogenannten Impulsblockade kommt.
Auch bei ALS wird ein deutlicher Anstieg des Jitters und der Instabilität einzelner Komponenten der PMU beobachtet. Dies erklärt sich dadurch, dass die durch die Keimung neu gebildeten Terminals und unreifen Synapsen nicht zuverlässig arbeiten. In diesem Fall werden Jitter und Impulsblockaden bei Patienten mit schnellem Fortschreiten des Prozesses am stärksten ausgeprägt.
Makroelektromyographie
Die Makroelektromyographie ermöglicht die Beurteilung der Größe der motorischen Einheiten in der Skelettmuskulatur. Bei der Untersuchung werden zwei Nadelelektroden gleichzeitig verwendet: eine spezielle Makroelektrode, die tief in den Muskel eingeführt wird, sodass sich die seitliche Abduktorfläche der Elektrode in der Muskeldicke befindet, und eine reguläre konzentrische Elektrode, die unter die Haut eingeführt wird. Die Makroelektromyographie basiert auf der Untersuchung des von einer Makroelektrode mit großer Abduktorfläche aufgezeichneten Potenzials.
Als Referenzelektrode dient eine konventionelle konzentrische Elektrode, die in einem Abstand von mindestens 30 cm von der Hauptmakroelektrode unter die Haut in die Zone minimaler Aktivität des zu untersuchenden Muskels, also möglichst weit vom motorischen Punkt des Muskels entfernt, eingeführt wird.
Eine weitere in der Kanüle montierte Elektrode zur Aufzeichnung von Potenzialen einzelner Muskelfasern registriert das Potenzial der Muskelfaser der untersuchten MU, das als Auslöser für die Mittelung des Makropotenzials dient. Das Signal von der Kanüle der Hauptelektrode gelangt ebenfalls in den Mittelwertbildner. 130–200 Impulse werden gemittelt (Epoche von 80 ms, ein Zeitraum von 60 ms wird für die Analyse verwendet), bis eine stabile Isolinie und ein Makropotenzial mit stabiler Amplitude der MU erscheinen. Die Registrierung erfolgt auf zwei Kanälen: Auf einem wird ein Signal von einer Muskelfaser der untersuchten MU aufgezeichnet, das die Mittelung auslöst, auf dem anderen wird das Signal zwischen Haupt- und Referenzelektrode wiedergegeben.
Der Hauptparameter zur Bewertung des Makropotentials der motorischen Einheit ist dessen Amplitude, gemessen von Spitze zu Spitze. Die Dauer des Potentials spielt bei dieser Methode keine Rolle. Es ist möglich, den Bereich der Makropotentiale der motorischen Einheit zu bewerten. Normalerweise gibt es eine große Bandbreite an Amplitudenwerten, mit zunehmendem Alter nimmt sie etwas zu. Bei neurogenen Erkrankungen nimmt die Amplitude der Makropotentiale der motorischen Einheit mit dem Grad der Reinnervation des Muskels zu. Bei neuronalen Erkrankungen ist sie am höchsten.
In den späten Stadien der Erkrankung nimmt die Amplitude der MU-Makropotentiale ab, insbesondere bei einer deutlichen Abnahme der Muskelkraft, die mit einer Abnahme der durch die Standard-Nadel-Elektromyographie aufgezeichneten MU-Parameter einhergeht.
Bei Myopathien ist eine Abnahme der Amplitude der Makropotentiale der motorischen Einheiten zu beobachten. Bei einigen Patienten sind die Durchschnittswerte jedoch normal, dennoch wird eine gewisse Anzahl von Potentialen mit reduzierter Amplitude beobachtet. Keine der Studien, die die Muskeln von Patienten mit Myopathie untersuchten, zeigte eine Zunahme der durchschnittlichen Amplitude der Makropotentiale der motorischen Einheiten.
Die Methode der Makroelektromyographie ist sehr arbeitsintensiv und hat daher in der Routinepraxis keine breite Anwendung gefunden.
Scanning-Elektromyographie
Die Methode ermöglicht die Untersuchung der zeitlichen und räumlichen Verteilung der elektrischen Aktivität der motorischen Einheit durch Scannen, d. h. schrittweise Bewegung der Elektrode im Bereich der Fasern der untersuchten motorischen Einheit. Die Scan-Elektromyographie liefert Informationen über die räumliche Lage der Muskelfasern im gesamten Raum der motorischen Einheit und kann indirekt auf das Vorhandensein von Muskelgruppen hinweisen, die durch den Prozess der Denervierung von Muskelfasern und deren wiederholte Reinnervierung gebildet werden.
Bei minimaler willkürlicher Muskelspannung dient die in den Muskel eingeführte Elektrode zur Aufzeichnung einer einzelnen Muskelfaser als Auslöser und mit Hilfe der konzentrischen Ausgangsnadelelektrode (Scanning-Elektrode) wird die PMU von allen Seiten mit einem Durchmesser von 50 mm aufgezeichnet. Die Methode basiert auf dem langsamen, schrittweisen Eintauchen einer Standardnadelelektrode in den Muskel, der Sammlung von Informationen über die Änderung der Potenzialparameter einer bestimmten MU und der Erstellung eines entsprechenden Bildes auf dem Monitorbildschirm. Die Scanning-Elektromyographie ist eine Reihe untereinander angeordneter Oszillogramme, die jeweils die an einem bestimmten Punkt aufgezeichneten und von der Ausgangsfläche der konzentrischen Nadelelektrode erfassten Biopotenzialschwingungen widerspiegeln.
Die anschließende Computeranalyse all dieser MUAPs und die Analyse ihrer dreidimensionalen Verteilung liefert Einblicke in das elektrophysiologische Profil von Motoneuronen.
Bei der Analyse der Daten der Rasterelektromyographie werden die Anzahl der Hauptspitzen des MUAP, ihre zeitliche Verschiebung ihres Auftretens und die Dauer der Intervalle zwischen dem Auftreten einzelner Bruchteile des Potenzials einer bestimmten MU bewertet und der Durchmesser der Faserverteilungszone in jeder der untersuchten MU berechnet.
Bei der DRP nehmen Amplitude und Dauer sowie der Bereich potenzieller Schwingungen in der Rasterelektromyographie zu. Der Querschnitt der Faserverteilungszone einzelner DE ändert sich jedoch nicht signifikant. Auch die für einen bestimmten Muskel charakteristische Anzahl der Fraktionen ändert sich nicht.
Kontraindikationen für das Verfahren
Es gibt praktisch keine Kontraindikationen für die Nadelelektromyographie. Als Einschränkung gilt der unbewusste Zustand des Patienten, wenn er den Muskel nicht willentlich anspannen kann. Selbst in diesem Fall ist es jedoch möglich, das Vorhandensein oder Fehlen des Stromprozesses in den Muskeln (anhand der spontanen Aktivität der Muskelfasern) festzustellen. Die Nadelelektromyographie sollte bei Muskeln mit ausgeprägten eitrigen Wunden, nicht heilenden Geschwüren und tiefen Verbrennungen mit Vorsicht durchgeführt werden.
Normale Leistung
Das DE ist ein strukturelles und funktionelles Element der Skelettmuskulatur. Es besteht aus einem Motoneuron im Vorderhorn der grauen Substanz des Rückenmarks, dessen Axon als myelinierte Nervenfaser aus der motorischen Wurzel austritt, und einer Gruppe von Muskelfasern, die über eine Synapse Kontakt mit den zahlreichen, der Myelinscheide beraubten Ästen dieses Axons – den Endstellen – herstellen.
Jede Muskelfaser eines Muskels hat ein eigenes Terminal, ist Teil nur einer motorischen Einheit und besitzt eine eigene Synapse. Axone beginnen sich einige Zentimeter vor dem Muskel intensiv zu verzweigen, um die Innervation jeder Muskelfaser dieser motorischen Einheit zu gewährleisten. Das Motoneuron erzeugt einen Nervenimpuls, der entlang des Axons weitergeleitet, in der Synapse verstärkt und zur Kontraktion aller zu dieser motorischen Einheit gehörenden Muskelfasern führt. Das während einer solchen Kontraktion der Muskelfasern aufgezeichnete gesamte bioelektrische Potenzial wird als motorisches Einheitspotenzial bezeichnet.
Motorische Einheitenpotentiale
Die Beurteilung des Zustands der motorischen Einheiten der menschlichen Skelettmuskulatur erfolgt anhand der Analyse der Parameter der von ihnen erzeugten Potenziale: Dauer, Amplitude und Form. Jede motorische Einheit entsteht durch die algebraische Addition der Potenziale aller Muskelfasern, die die motorische Einheit bilden, die als Einheit fungiert.
Wenn sich die Erregungswelle entlang der Muskelfasern zur Elektrode ausbreitet, erscheint auf dem Monitorbildschirm ein dreiphasiges Potenzial: Die erste Abweichung ist positiv, dann folgt eine schnelle negative Spitze, und das Potenzial endet mit einer dritten, ebenfalls positiven Abweichung. Diese Phasen können unterschiedliche Amplituden, Dauern und Flächen haben, die von der Position der Ausgangsoberfläche der Elektrode im Verhältnis zum zentralen Teil des aufgezeichneten DE abhängen.
Die Parameter der PMU spiegeln die Größe des DE, die Menge, die gegenseitige Anordnung der Muskelfasern und die Dichte ihrer Verteilung in jedem spezifischen DE wider.
Normale Potenzialdauer einer motorischen Einheit
Der Hauptparameter der PDE ist ihre Dauer oder Länge, gemessen als Zeit in Millisekunden vom Beginn der Signalabweichung von der Mittellinie bis zu ihrer vollständigen Rückkehr dorthin.
Die Dauer der PMU bei einem gesunden Menschen hängt von Muskel und Alter ab. Mit zunehmendem Alter nimmt die Dauer der PMU zu. Um einheitliche Kriterien für die Normierung der PMU-Studie zu schaffen, wurden spezielle Tabellen mit durchschnittlichen Normaldauern für verschiedene Muskeln unterschiedlichen Alters entwickelt. Ein Auszug dieser Tabellen ist unten dargestellt.
Als Maßstab für die Beurteilung des MU-Zustands im Muskel dient die durchschnittliche Dauer von 20 verschiedenen MUAPs, die an verschiedenen Punkten des untersuchten Muskels aufgezeichnet wurden. Der während der Untersuchung ermittelte Durchschnittswert wird mit dem entsprechenden Indikator in der Tabelle verglichen und die Abweichung von der Norm (in Prozent) berechnet. Die durchschnittliche Dauer der MUAP gilt als normal, wenn sie innerhalb der Grenzen von ±12 % des in der Tabelle angegebenen Wertes liegt (im Ausland gilt die durchschnittliche Dauer der MUAP als normal, wenn sie innerhalb der Grenzen von ±20 % liegt).
[ 14 ], [ 15 ], [ 16 ], [ 17 ]
Dauer motorischer Einheitenpotentiale in der Pathologie
Das Hauptmuster der Veränderungen in der Dauer der PDE bei pathologischen Zuständen besteht darin, dass sie bei neurogenen Erkrankungen zunimmt und bei synaptischen und primären Muskelpathologien abnimmt.
Um den Grad der PMU-Veränderung in Muskeln mit verschiedenen Läsionen des peripheren neuromotorischen Apparats genauer beurteilen zu können, wird für jeden Muskel ein Histogramm der PMU-Verteilung nach Dauer verwendet, da deren Durchschnittswert bei offensichtlicher Muskelpathologie im Normbereich liegen kann. Normalerweise hat das Histogramm die Form einer Normalverteilung, deren Maximum mit der durchschnittlichen PMU-Dauer für einen bestimmten Muskel übereinstimmt. Bei jeder Pathologie des peripheren neuromotorischen Apparats verändert sich die Form des Histogramms signifikant.
Elektromyographische Stadien des pathologischen Prozesses
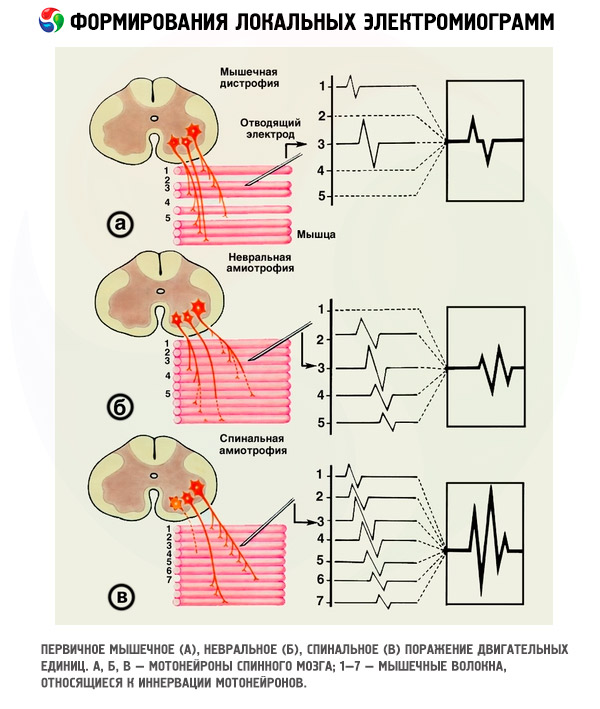
Basierend auf der Änderung der Dauer der MU bei Erkrankungen der Motoneuronen des Rückenmarks, wenn alle in den Muskeln auftretenden Änderungen in einem relativ kurzen Zeitraum verfolgt werden können, wurden sechs EMG-Stadien identifiziert, die die allgemeinen Muster der Umstrukturierung der MU während des Denervierungs-Reinnervierungsprozesses (DRP) vom Ausbruch der Krankheit bis zum fast vollständigen Absterben des Muskels widerspiegeln.
Alle neurogenen Erkrankungen sind durch das Absterben einer größeren oder geringeren Anzahl von Motoneuronen oder deren Axonen gekennzeichnet. Die überlebenden Motoneuronen innervieren die „fremden“ Muskelfasern, die ihrer nervlichen Kontrolle beraubt sind, und erhöhen dadurch ihre Zahl in ihren MU. In der Elektromyographie manifestiert sich dieser Prozess durch einen allmählichen Anstieg der Potenzialparameter solcher MU. Der gesamte Veränderungszyklus im Histogramm der Verteilung der MU nach Dauer bei neuronalen Erkrankungen wird üblicherweise in fünf EMG-Stadien unterteilt, die den Prozess der kompensatorischen Innervation in den Muskeln widerspiegeln. Obwohl diese Einteilung üblich ist, hilft sie, alle Stadien der Entwicklung von DRP in jedem spezifischen Muskel zu verstehen und nachzuverfolgen, da jedes Stadium eine bestimmte Phase der Reinnervation und ihren Schweregrad widerspiegelt. Es ist unangemessen, Stadium VI als Histogramm darzustellen, da es den Endpunkt des „umgekehrten“ Prozesses widerspiegelt, d. h. den Prozess der Dekompensation und Zerstörung der Muskel-MU.
Unter Fachärzten in unserem Land haben sich diese Stadien bei der Diagnose verschiedener neuromuskulärer Erkrankungen durchgesetzt. Sie sind im Computerprogramm von Haushaltselektromyographen enthalten, das die automatische Erstellung von Histogrammen ermöglicht, die das Stadium des Prozesses anzeigen. Eine Veränderung des Stadiums in die eine oder andere Richtung bei einer wiederholten Untersuchung des Patienten zeigt die weiteren Aussichten für die Entwicklung von DRP.
- Stadium I: Die durchschnittliche Dauer des MUAP ist um 13–20 % reduziert. Dieses Stadium spiegelt die allererste Phase der Erkrankung wider, wenn die Denervierung bereits begonnen hat und der Prozess der Reinnervierung elektromyographisch noch nicht manifestiert ist. Einige denervierte Muskelfasern, denen aufgrund einer Pathologie des Motoneurons oder seines Axons der Impulseinfluss entzogen ist, fallen aus der Zusammensetzung einiger MU heraus. Die Anzahl der Muskelfasern in solchen MUAPs nimmt ab, was zu einer Verkürzung der Dauer einzelner Potenziale führt. Im Stadium I treten eine bestimmte Anzahl schmalerer Potenziale als in gesunder Muskulatur auf, was eine leichte Verkürzung der durchschnittlichen Dauer bewirkt. Das Histogramm der MUAP-Verteilung beginnt sich nach links in Richtung kleinerer Werte zu verschieben.
- Stadium II: Die durchschnittliche Dauer des MUAP wird um 21 % oder mehr reduziert. Bei DRP wird dieses Stadium äußerst selten und nur in Fällen beobachtet, in denen aus irgendeinem Grund keine Reinnervation stattfindet oder durch einen Faktor (z. B. Alkohol, Strahlung usw.) unterdrückt wird, während die Denervierung im Gegenteil zunimmt und es zu einem massiven Absterben von Muskelfasern im MUAP kommt. Dies führt dazu, dass die meisten oder fast alle MUAPs kürzer als normal dauern, wodurch die durchschnittliche Dauer weiter abnimmt. Das Histogramm der MUAP-Verteilung verschiebt sich signifikant in Richtung kleinerer Werte. Die Stadien I-II spiegeln Veränderungen in den MUAPs wider, die durch eine Abnahme der Anzahl funktionierender Muskelfasern in ihnen verursacht werden.
- Stadium III: Die durchschnittliche Dauer des MUAP liegt für einen bestimmten Muskel innerhalb von ± 20 % der Norm. Dieses Stadium ist durch das Auftreten einer bestimmten Anzahl von Potenzialen mit erhöhter Dauer gekennzeichnet, die normalerweise nicht erkennbar sind. Das Auftreten dieser MUAPs zeigt den Beginn der Reinnervierung an, d. h. denervierte Muskelfasern werden in andere MUAPs einbezogen, wodurch die Parameter ihrer Potenziale zunehmen. Im Muskel werden gleichzeitig MUAPs sowohl mit verringerter und normaler als auch mit erhöhter Dauer aufgezeichnet, die Anzahl vergrößerter MUAPs im Muskel variiert von einem bis zu mehreren. Die durchschnittliche Dauer des MUAP in Stadium III kann normal sein, aber das Erscheinungsbild des Histogramms weicht von der Norm ab. Es hat nicht die Form einer Normalverteilung, sondern ist „abgeflacht“, gestreckt und beginnt sich nach rechts in Richtung höherer Werte zu verschieben. Es wird vorgeschlagen, Stadium III in zwei Untergruppen zu unterteilen – IIIA und IIIB. Sie unterscheiden sich lediglich darin, dass im Stadium IIIA die durchschnittliche Dauer des MUAP um 1–20 % reduziert ist und im Stadium IIIB entweder vollständig dem durchschnittlichen Normwert entspricht oder um 1–20 % erhöht ist. Im Stadium IIIB werden etwas mehr MUAPs mit erhöhter Dauer registriert als im Stadium IIIA. Die Praxis hat gezeigt, dass eine solche Aufteilung des dritten Stadiums in zwei Untergruppen keine große Bedeutung hat. Tatsächlich bedeutet Stadium III lediglich das Auftreten der ersten EMG-Anzeichen einer Reinnervation im Muskel.
- Stadium IV: Die durchschnittliche Dauer des MUAP erhöht sich um 21–40 %. Dieses Stadium ist durch eine Erhöhung der durchschnittlichen Dauer des MUAP gekennzeichnet, da neben normalen MUAPs eine große Anzahl von Potenzialen mit erhöhter Dauer auftritt. MUAPs mit reduzierter Dauer werden in diesem Stadium äußerst selten erfasst. Das Histogramm ist nach rechts zu größeren Werten verschoben, seine Form ist unterschiedlich und hängt vom Verhältnis der MUAPs mit normaler und erhöhter Dauer ab.
- Stadium V: Die durchschnittliche Dauer des MUAP ist um 41 % oder mehr erhöht. Dieses Stadium ist durch das Vorhandensein überwiegend großer und „riesiger“ MUAPs gekennzeichnet, während MUAPs normaler Dauer praktisch fehlen. Das Histogramm ist deutlich nach rechts verschoben, gestreckt und in der Regel offen. Dieses Stadium spiegelt das maximale Volumen der Reinnervation im Muskel sowie deren Effektivität wider: Je mehr riesige MUAPs vorhanden sind, desto effektiver ist die Reinnervation.
- Stadium VI: Die durchschnittliche Dauer des MUAP liegt im Normbereich oder ist um mehr als 12 % reduziert. Dieses Stadium ist durch das Vorhandensein von MUAPs (Potenziale sich verschlechternder MUs) gekennzeichnet, deren Form verändert ist. Ihre Parameter können formal normal oder reduziert sein, die Form der MUAPs ist jedoch verändert: Die Potenziale weisen keine scharfen Spitzen auf, sind gestreckt, abgerundet, die Potenzialanstiegszeit ist stark erhöht. Dieses Stadium wird im letzten Stadium der Dekompensation des DRP beobachtet, wenn die meisten Motoneuronen des Rückenmarks bereits abgestorben sind und der Rest intensiv abstirbt. Die Dekompensation des Prozesses beginnt in dem Moment, in dem der Denervierungsprozess zunimmt und die Innervationsquellen immer weniger werden. Im EMG ist das Dekompensationsstadium durch die folgenden Anzeichen gekennzeichnet: Die MUAP-Parameter beginnen abzunehmen, riesige MUAPs verschwinden allmählich, die Intensität des PF nimmt stark zu, riesige POWs treten auf, was auf das Absterben vieler benachbarter Muskelfasern hinweist. Diese Anzeichen deuten darauf hin, dass die Motoneuronen in diesem Muskel aufgrund funktioneller Insuffizienz ihre Sprossungskapazität erschöpft haben und ihre Fasern nicht mehr vollständig steuern können. Infolgedessen nimmt die Anzahl der Muskelfasern in der motorischen Einheit progressiv ab, die Impulsleitungsmechanismen werden gestört, die Potenziale dieser motorischen Einheiten werden gerundet, ihre Amplitude nimmt ab und ihre Dauer verkürzt sich. Die Erstellung eines Histogramms ist in diesem Stadium des Prozesses nicht sinnvoll, da es, ebenso wie die durchschnittliche Dauer der PMU, nicht mehr den tatsächlichen Zustand des Muskels widerspiegelt. Das Hauptsymptom von Stadium VI ist eine Veränderung der Form aller PMUs.
EMG-Stadien werden nicht nur bei neurogenen Erkrankungen, sondern auch bei verschiedenen primären Muskelerkrankungen verwendet, um die Tiefe der Muskelpathologie zu charakterisieren. In diesem Fall spiegelt das EMG-Stadium nicht den DRP, sondern den Schweregrad der Pathologie wider und wird als „EMG-Stadium des pathologischen Prozesses“ bezeichnet. Bei primären Muskeldystrophien können stark polyphasische PMUs mit länger andauernden Satelliten auftreten, was ihren Durchschnittswert signifikant erhöht und dem III. oder sogar IV. EMG-Stadium des pathologischen Prozesses entspricht.
Diagnostische Bedeutung der EMG-Stadien
- Bei neuronalen Erkrankungen werden beim gleichen Patienten häufig unterschiedliche EMG-Stadien in unterschiedlichen Muskeln nachgewiesen – die Stadien III bis VI werden nur sehr selten – und zwar ganz am Anfang der Erkrankung und nur in einzelnen Muskeln.
- Bei axonalen und demyelinisierenden Erkrankungen werden am häufigsten die Stadien III und IV festgestellt, seltener die Stadien I und II. Von Stadium V spricht man, wenn in einzelnen, am stärksten betroffenen Muskeln eine erhebliche Anzahl Axone absterben.
- Bei primären Muskelerkrankungen kommt es aufgrund einer Muskelpathologie zu einem Verlust von Muskelfasern aus der Zusammensetzung der MU: einer Verringerung des Durchmessers der Muskelfasern, ihrer Spaltung, Fragmentierung oder anderen Schäden, die die Anzahl der Muskelfasern in der MU verringern oder das Muskelvolumen reduzieren. All dies führt zu einer Verkürzung der Dauer der PMU. Daher werden bei den meisten primären Muskelerkrankungen und Myasthenie die Stadien I und II nachgewiesen, bei Polymyositis zunächst nur I und I und nach der Genesung die Stadien III und sogar IV.
Potentialamplitude der motorischen Einheit
Die Amplitude ist ein zusätzlicher, aber sehr wichtiger Parameter bei der Analyse des MUAP. Sie wird „von Spitze zu Spitze“ gemessen, d. h. vom tiefsten Punkt des positiven bis zum höchsten Punkt des negativen Peaks. Bei der Anzeige der MUAP auf dem Bildschirm wird deren Amplitude automatisch ermittelt. Sowohl die durchschnittliche als auch die maximale Amplitude des im untersuchten Muskel erfassten MUAP werden ermittelt.
Die Durchschnittswerte der MUAP-Amplitude in den proximalen Muskeln gesunder Menschen liegen in den meisten Fällen bei 500–600 µV, in den distalen Muskeln bei 600–800 µV, während die maximale Amplitude 1500–1700 µV nicht überschreitet. Diese Werte sind sehr bedingt und können teilweise variieren. Bei Kindern im Alter von 8–12 Jahren liegt die durchschnittliche MUAP-Amplitude in der Regel zwischen 300 und 400 µV und das Maximum überschreitet 800 µV nicht; bei älteren Kindern liegen diese Werte bei 500 bzw. 1000 µV. In den Gesichtsmuskeln ist die MUAP-Amplitude deutlich geringer.
Bei Sportlern wird eine erhöhte Amplitude des MUAP in trainierten Muskeln festgestellt. Folglich kann eine Erhöhung der durchschnittlichen Amplitude des MUAP in den Muskeln gesunder Sportler nicht als pathologisch angesehen werden, da sie auf die Umstrukturierung des MU aufgrund längerer Muskelbelastung zurückzuführen ist.
Bei allen neurogenen Erkrankungen nimmt die Amplitude des PMU in der Regel mit zunehmender Dauer zu: Je länger das Potenzial anhält, desto höher ist seine Amplitude.
Die deutlichste Zunahme der MUAP-Amplitude wird bei neuronalen Erkrankungen wie spinaler Amyotrophie und den Folgen einer Poliomyelitis beobachtet. Sie dient als zusätzliches Kriterium zur Diagnose neurogener Muskelerkrankungen. Die Zunahme der MUAP-Amplitude wird durch die Reorganisation der MU im Muskel, eine Zunahme der Muskelfaseranzahl im Elektrodenleitungsbereich, die Synchronisation ihrer Aktivität und eine Vergrößerung des Muskelfaserdurchmessers verursacht.
Bei einigen primären Muskelerkrankungen wie Polymyositis, primärer Muskeldystrophie, dystrophischer Myotonie usw. wird manchmal eine Erhöhung sowohl der durchschnittlichen als auch der maximalen Amplitude des MUAP beobachtet.
Wellenform des Motoreinheitspotentials
Die Form der PDE hängt von der Struktur der DE, dem Grad der Synchronisation der Potentiale ihrer Muskelfasern, der Position der Elektrode im Verhältnis zu den Muskelfasern der analysierten DE und deren Innervationszonen ab. Die Form des Potentials hat keinen diagnostischen Wert.
In der klinischen Praxis wird die Form des MUAP anhand der Anzahl der Phasen und/oder Windungen im Potenzial analysiert. Jede positiv-negative Potenzialabweichung, die die Isolinie erreicht und kreuzt, wird als Phase bezeichnet, und eine positiv-negative Potenzialabweichung, die die Isolinie nicht erreicht, wird als Windung bezeichnet.
Ein Potenzial gilt als mehrphasig, wenn es fünf oder mehr Phasen aufweist und die Achsenlinie mindestens viermal kreuzt. Das Potenzial kann zusätzliche Windungen aufweisen, die die Achsenlinie nicht kreuzen. Windungen können sowohl im negativen als auch im positiven Teil des Potenzials vorhanden sein.
In den Muskeln gesunder Menschen werden die MUAPs normalerweise durch dreiphasige Potentialschwingungen dargestellt. Bei der Aufzeichnung des MUAP in der Endplattenzone kann es jedoch zwei Phasen aufweisen und seinen anfänglichen positiven Teil verlieren.
Normalerweise überschreitet die Anzahl polyphasischer MUAPs nicht 5–15 %. Eine Zunahme polyphasischer MUAPs gilt als Zeichen einer Störung der MU-Struktur aufgrund eines pathologischen Prozesses. Polyphasische und pseudopolyphasische MUAPs werden sowohl bei neuronalen und axonalen Erkrankungen als auch bei primären Muskelerkrankungen beobachtet.
[ 29 ], [ 30 ], [ 31 ], [ 32 ], [ 33 ]
Spontane Aktivität
Unter normalen Bedingungen, wenn die Elektrode im entspannten Muskel eines gesunden Menschen stationär ist, tritt keine elektrische Aktivität auf. In pathologischen Fällen tritt eine spontane Aktivität der Muskelfasern oder eine DE auf. Die spontane Aktivität hängt nicht vom Willen des Patienten ab, er kann sie nicht stoppen oder willkürlich auslösen.
Spontane Aktivität der Muskelfasern
Die spontane Aktivität von Muskelfasern umfasst Fibrillationspotentiale (FP) und positive scharfe Wellen (PSW). FP und PSB werden ausschließlich unter pathologischen Bedingungen aufgezeichnet, wenn eine konzentrische Nadelelektrode in den Muskel eingeführt wird. FP ist das Potential einer Muskelfaser, PSB ist eine langsame Schwingung, die nach einer schnellen positiven Auslenkung ohne scharfen negativen Peak auftritt. PSB spiegelt die Beteiligung sowohl einer als auch mehrerer benachbarter Fasern wider.
Die Untersuchung der spontanen Aktivität von Muskelfasern unter den Bedingungen einer klinischen Untersuchung eines Patienten ist die bequemste elektrophysiologische Methode, die es ermöglicht, den Grad der Vollständigkeit und Stabilität der Nerveneinwirkungen auf die Muskelfasern eines Skelettmuskels in seiner Pathologie zu beurteilen.
Spontane Muskelfaseraktivität kann bei allen Erkrankungen des peripheren neuromotorischen Apparats auftreten. Bei neurogenen Erkrankungen sowie bei Synapsenerkrankungen (Myasthenie und myasthenische Syndrome) spiegelt die spontane Muskelfaseraktivität den Prozess ihrer Denervierung wider. Bei den meisten primären Muskelerkrankungen spiegelt die spontane Muskelfaseraktivität eine Schädigung der Muskelfasern (deren Spaltung, Fragmentierung etc.) sowie deren durch den Entzündungsprozess verursachte Pathologie (bei entzündlichen Myopathien – Polymyositis, Dermatomyositis) wider. In beiden Fällen weisen PF und POV auf einen laufenden Prozess im Muskel hin; sie werden normalerweise nie erfasst.
- Die Dauer der PF beträgt 1–5 ms (sie hat keinen diagnostischen Wert), und die Amplitude schwankt in sehr weiten Grenzen (durchschnittlich 118 ± 114 μV). Gelegentlich werden auch PF mit hoher Amplitude (bis zu 2000 μV) nachgewiesen, meist bei Patienten mit chronischen Erkrankungen. Der Zeitpunkt des Auftretens der PF hängt vom Ort der Nervenschädigung ab. In den meisten Fällen treten sie 7–20 Tage nach der Denervierung auf.
- Wenn aus irgendeinem Grund die Reinnervation der denervierten Muskelfaser nicht erfolgt, stirbt sie mit der Zeit ab und erzeugt POWs, die vom EMG als Zeichen des Absterbens der denervierten Muskelfaser gewertet werden, die nicht die Innervation erhalten hat, die sie zuvor verloren hatte. Anhand der Anzahl der in jedem Muskel aufgezeichneten PFs und POWs können Grad und Tiefe seiner Denervierung oder das Volumen abgestorbener Muskelfasern indirekt beurteilt werden. Die Dauer der POW beträgt 1,5 bis 70 ms (in den meisten Fällen bis zu 10 ms). Die sogenannten riesigen POWs, die länger als 20 ms andauern, werden bei längerer Denervierung einer großen Anzahl benachbarter Muskelfasern sowie bei Polymyositis festgestellt. Die Amplitude der POW schwankt üblicherweise zwischen 10 und 1800 μV. POWs mit großer Amplitude und Dauer werden häufiger in späteren Stadien der Denervierung festgestellt („riesige“ POWs). POVs werden erstmals 16–30 Tage nach dem ersten Auftreten der PF registriert; sie können nach der Denervierung mehrere Jahre im Muskel persistieren. Bei Patienten mit entzündlichen Läsionen der peripheren Nerven werden POVs in der Regel später erkannt als bei Patienten mit traumatischen Läsionen.
PF und POV reagieren am schnellsten auf den Beginn der Therapie: Bei Wirksamkeit nimmt der Schweregrad von PF und POV nach 2 Wochen ab. Im Gegenteil, wenn die Behandlung unwirksam oder unzureichend wirksam ist, nimmt ihr Schweregrad zu, was es ermöglicht, die Analyse von PF und POV als Indikator für die Wirksamkeit der verwendeten Medikamente zu verwenden.
Myotone und pseudomyotone Entladungen
Myotone und pseudomyotone Entladungen oder Hochfrequenzentladungen beziehen sich ebenfalls auf die spontane Aktivität von Muskelfasern. Myotone und pseudomyotone Entladungen unterscheiden sich in einer Reihe von Merkmalen, wobei das wichtigste die hohe Wiederholbarkeit der Elemente, aus denen die Entladung besteht, d. h. die hohe Frequenz der Potentiale in der Entladung ist. Der Begriff „pseudomyotone Entladung“ wird zunehmend durch den Begriff „Hochfrequenzentladung“ ersetzt.
- Myotone Entladungen sind ein Phänomen, das bei Patienten mit verschiedenen Formen der Myotonie beobachtet wird. Beim Hören ähnelt es dem Klang eines Sturzkampfbombers. Auf dem Bildschirm erscheinen diese Entladungen als sich wiederholende Potentiale mit allmählich abnehmender Amplitude und zunehmend zunehmenden Intervallen (was zu einer Abnahme der Tonhöhe führt). Myotone Entladungen werden manchmal bei bestimmten Formen endokriner Erkrankungen (z. B. Hypothyreose) beobachtet. Myotone Entladungen treten entweder spontan oder nach einer leichten Kontraktion oder mechanischen Reizung des Muskels durch Einführen einer Nadelelektrode oder durch einfaches Klopfen auf den Muskel auf.
- Pseudomyotone Entladungen (hochfrequente Entladungen) werden bei einigen neuromuskulären Erkrankungen registriert, sowohl in Verbindung mit als auch ohne Denervierung von Muskelfasern. Sie gelten als Folge einer ephaptischen Erregungsübertragung mit verminderter isolierender Wirkung der Muskelfasermembran, wodurch die Voraussetzung für die Erregungsausbreitung von einer Faser auf die benachbarte geschaffen wird: Der Schrittmacher einer der Fasern gibt den Impulsrhythmus vor, der den benachbarten Fasern aufgezwungen wird und die einzigartige Form der Komplexe bewirkt. Entladungen beginnen und enden plötzlich. Ihr Hauptunterschied zu myotonen Entladungen ist das Fehlen eines Amplitudenabfalls der Komponenten. Pseudomyotone Entladungen werden bei verschiedenen Formen von Myopathie, Polymyositis, Denervierungssyndromen (in den späten Stadien der Reinnervation), spinalen und neuralen Amyotrophien (Charcot-Marie-Tooth-Krankheit), endokrinen Pathologien, Nervenverletzungen oder -kompressionen und einigen anderen Erkrankungen beobachtet.
Spontane motorische Einheitsaktivität
Die spontane Aktivität der motorischen Einheit wird durch Faszikulationspotentiale repräsentiert. Faszikulationen sind spontane Kontraktionen der gesamten motorischen Einheit, die bei völlig entspanntem Muskel auftreten. Ihr Auftreten ist mit Motoneuronerkrankungen, der Überlastung der Muskelfasern, der Reizung einzelner Muskelabschnitte sowie funktionellen und morphologischen Umstrukturierungen verbunden.
Das Auftreten multipler Faszikulationspotentiale in der Muskulatur gilt als eines der Hauptsymptome einer Schädigung der Motoneuronen des Rückenmarks. Eine Ausnahme bilden „gutartige“ Faszikulationspotentiale, die manchmal bei Patienten beobachtet werden, die über ständige Muskelzuckungen klagen, aber keine Muskelschwäche oder andere Symptome bemerken.
Einzelne Faszikulationspotentiale können auch bei neurogenen und sogar primären Muskelerkrankungen wie Myotonie, Polymyositis, endokrinen, metabolischen und mitochondrialen Myopathien nachgewiesen werden.
Es wurden Faszikulationspotentiale beschrieben, die bei Hochleistungssportlern nach anstrengender körperlicher Betätigung auftreten. Sie können auch bei gesunden, aber leicht erregbaren Menschen, bei Patienten mit Tunnelsyndromen, Polyneuropathien und bei älteren Menschen auftreten. Im Gegensatz zu Motoneuronerkrankungen ist ihre Anzahl im Muskel jedoch sehr gering, und die Parameter sind in der Regel normal.
Die Parameter der Faszikulationspotentiale (Amplitude und Dauer) entsprechen den Parametern des in einem bestimmten Muskel aufgezeichneten MUAP und können sich während der Entwicklung der Krankheit parallel zu den Änderungen des MUAP ändern.
Nadelelektromyographie in der Diagnostik von Erkrankungen der Motoneuronen des Rückenmarks und der peripheren Nerven
Jede neurogene Pathologie wird von DRP begleitet, deren Schweregrad vom Grad der Schädigung der Innervationsquellen und von der Ebene des peripheren neuromotorischen Apparats – neuronal oder axonal – abhängt, auf der die Schädigung aufgetreten ist. In beiden Fällen wird die verlorene Funktion durch die überlebenden Nervenfasern wiederhergestellt, und diese beginnen sich intensiv zu verzweigen und bilden zahlreiche Triebe, die auf die denervierten Muskelfasern gerichtet sind. Diese Verzweigung wird in der Literatur als „Sprossen“ bezeichnet.
Es gibt zwei Hauptarten der Sprossung: kollaterale und terminale. Kollaterale Sprossung bezeichnet die Verzweigung von Axonen im Bereich der Ranvierschen Knoten, terminale Sprossung bezeichnet die Verzweigung des letzten, unmyelinierten Axonabschnitts. Es wurde nachgewiesen, dass die Art der Sprossung von der Art des Faktors abhängt, der die Störung der Nervenkontrolle verursacht hat. Beispielsweise erfolgt bei einer Botulinumintoxikation die Verzweigung ausschließlich in der Terminalzone, und bei einer chirurgischen Denervierung treten sowohl terminale als auch kollaterale Sprossungen auf.
In der Elektromyographie sind diese Zustände der MU in verschiedenen Stadien des Reinnervationsprozesses durch das Auftreten von MUAP mit erhöhter Amplitude und Dauer gekennzeichnet. Eine Ausnahme bilden die Anfangsstadien der bulbären Form der ALS, in denen die MUAP-Parameter mehrere Monate lang im Normbereich liegen.
Elektromyographische Kriterien für Erkrankungen der Motoneuronen des Rückenmarks
- Das Vorhandensein ausgeprägter Faszikulationspotentiale (das Hauptkriterium für eine Schädigung der Motoneuronen des Rückenmarks).
- Eine Erhöhung der PDE-Parameter und ihrer Polyphasie spiegelt die Schwere des Reinnervationsprozesses wider.
- Das Auftreten einer spontanen Aktivität der Muskelfasern in den Muskeln – PF und POV – weist auf das Vorhandensein eines laufenden Denervierungsprozesses hin.
Faszikulationspotentiale sind ein obligatorisches elektrophysiologisches Zeichen für eine Schädigung der Motoneuronen des Rückenmarks. Sie werden bereits in den frühesten Stadien des pathologischen Prozesses erkannt, noch bevor Anzeichen einer Denervierung auftreten.
Da neuronale Erkrankungen einen ständigen Prozess der Denervierung und Reinnervierung mit sich bringen, werden die Motoneuronen beim gleichzeitigen Absterben einer großen Anzahl von MUs immer größer, ihre Dauer und Amplitude nehmen zu. Das Ausmaß der Zunahme hängt von der Dauer und dem Stadium der Erkrankung ab.
Der Schweregrad von PF und POV hängt von der Schwere des pathologischen Prozesses und dem Grad der Muskeldenervierung ab. Bei schnell fortschreitenden Erkrankungen (z. B. ALS) finden sich PF und POV in den meisten Muskeln, bei langsam fortschreitenden Erkrankungen (einigen Formen spinaler Amyotrophien) nur in der Hälfte der Muskeln und beim Post-Poliomyelitis-Syndrom in weniger als einem Drittel.
Elektromyographische Kriterien für Erkrankungen peripherer Nervenaxone
Die Nadelelektromyographie in der Diagnostik peripherer Nervenerkrankungen ist eine zusätzliche, aber notwendige Untersuchungsmethode, um den Grad der Schädigung des vom betroffenen Nerv innervierten Muskels zu bestimmen. Die Studie ermöglicht die Klärung des Vorhandenseins von Anzeichen einer Denervierung (SF), des Ausmaßes des Muskelfaserverlusts im Muskel (Gesamtzahl der MUFs und Vorhandensein riesiger MUFs), des Schweregrads der Reinnervation und ihrer Wirksamkeit (Grad der Erhöhung der MUF-Parameter, maximale Amplitude der MUF im Muskel).
Die wichtigsten elektromyographischen Zeichen des Axonprozesses:
- Erhöhung des Durchschnittswerts der Amplitude der PDE;
- das Vorhandensein von PF und POV (mit aktueller Denervierung);
- Verlängerung der PDE-Dauer (der Durchschnittswert kann im Normbereich liegen, d. h. ±12 %);
- Polyphasie der PDE;
- einzelne Faszikulationspotentiale (nicht in jedem Muskel).
Bei Schäden an Axonen peripherer Nerven (verschiedene Polyneuropathien) tritt DRP ebenfalls auf, aber sein Schweregrad ist viel geringer als bei neuronalen Erkrankungen. Folglich sind MUAPs in viel geringerem Maße erhöht. Trotzdem gilt die Grundregel der MUAP-Veränderung bei neurogenen Erkrankungen auch für Schäden an Axonen motorischer Nerven (d. h. der Grad der Erhöhung der MUAP-Parameter und ihre Polyphasie hängen vom Grad der Nervenschädigung und der Schwere der Reinnervation ab). Eine Ausnahme bilden pathologische Zustände, die mit einem schnellen Tod von Axonen motorischer Nerven aufgrund eines Traumas (oder eines anderen pathologischen Zustands, der zum Tod einer großen Anzahl Axonen führt) einhergehen. In diesem Fall treten dieselben riesigen MUAPs (mit einer Amplitude von über 5000 μV) auf wie bei neuronalen Erkrankungen. Solche MUAPs werden bei Langzeitformen axonaler Pathologie, CIDP und neuralen Amyotrophien beobachtet.
Wenn bei axonalen Polyneuropathien zunächst die Amplitude des MUAP zunimmt, dann steigen im Demyelinisierungsprozess mit Verschlechterung des Funktionszustands des Muskels (Abnahme seiner Kraft) die Durchschnittswerte der MUAP-Dauer allmählich an; viel häufiger als im axonalen Prozess werden polyphasische MUAPs und Faszikulationspotentiale nachgewiesen und seltener PF und POV.
[ 37 ], [ 38 ], [ 39 ], [ 40 ], [ 41 ], [ 42 ]
Nadelelektromyographie in der Diagnostik synaptischer und primärer Muskelerkrankungen
Synaptische und primäre Muskelerkrankungen weisen typischerweise eine Verkürzung der durchschnittlichen Dauer der MUAP auf. Der Grad der Verkürzung der MUAP-Dauer korreliert mit der Kraftabnahme. In einigen Fällen liegen die MUAP-Parameter im Normbereich, bei PMD können sie sogar erhöht sein.